Debemos reconocer que la pandemia del Covid-19 está teniendo consecuencias que afectan a nuestra vida, nuestro trabajo, el mundo de la enseñanza, la cultura, el ocio, etc… En fin, afecta el día a día en que normalmente nos movemos. Tenemos que saber que Covid-19 es el nombre oficial que la OMS le dio en febrero de 2020 a la enfermedad infecciosa causada por el nuevo coronavirus, es decir por el virus SARS-CoV-2. Esta pandemia también está afectando a muchas personas, pequeños empresarios, autónomos, trabajadores e inmigrantes sin papeles, que han perdido su trabajo y, en muchos casos, sus ingresos. Por ello es tan importante encontrar soluciones farmacológicas que puedan paliar los efectos del virus. Distintos equipos investigadores están intentando  obtener cuanto antes antivirales para inhibir los contagios, mientras a más largo plazo se está tratando de encontrar una vacuna efectiva contra el virus. Hoy en día hay necesidad de adoptar un enfoque riguroso, multidisciplinar y coherente con la respuesta que den los distintos países y las instancias mundiales, como la OMS. En este artículo me centraré en los avances de la Inteligencia Artificial (IA) para encontrar soluciones más rápidas, potentes y efectivas para combatir a bacterias, que se han hecho resistentes a los actuales antibióticos, y virus, que puedan terminar siendo pandemias, así como otro tipo de enfermedades, como el cáncer. Ello es aún más importante cuanto tenemos miles de distintas bacterias y virus, así como múltiples otras enfermedades que pueden atacar a los seres humanos. El 12 de marzo de 2020, Will Douglas Heaven, de MIT News, explicó que se cree que la Inteligencia Artificial (IA) podría ayudar con la próxima pandemia, pero ya no con esta actual pandemia del Covid-19. Algunas cosas deben cambiar si queremos que la IA sea útil la próxima vez y, según Heaven, es posible que algunas no nos gusten. Fue un algoritmo de IA el que vio venir la pandemia por primera vez, o eso dice la historia. En efecto, el 30 de diciembre de 2019, una compañía de inteligencia artificial llamada BlueDot, que utiliza el aprendizaje automático para monitorear brotes de enfermedades infecciosas en todo el mundo, alertó a sus clientes, incluidos varios gobiernos, hospitales y empresas, sobre un aumento inusual en los casos de neumonía en Wuhan, China, que presagiaban alguna posible epidemia. Pero pasarían otros nueve días antes de que la Organización Mundial de la Salud (OMS) señalara oficialmente lo que todos conocemos como Covid-19. Ello demostró que la IA es capaz de detectar con gran rapidez y eficacia lo que los humanos tenemos mucha más dificultad de hacerlo.
obtener cuanto antes antivirales para inhibir los contagios, mientras a más largo plazo se está tratando de encontrar una vacuna efectiva contra el virus. Hoy en día hay necesidad de adoptar un enfoque riguroso, multidisciplinar y coherente con la respuesta que den los distintos países y las instancias mundiales, como la OMS. En este artículo me centraré en los avances de la Inteligencia Artificial (IA) para encontrar soluciones más rápidas, potentes y efectivas para combatir a bacterias, que se han hecho resistentes a los actuales antibióticos, y virus, que puedan terminar siendo pandemias, así como otro tipo de enfermedades, como el cáncer. Ello es aún más importante cuanto tenemos miles de distintas bacterias y virus, así como múltiples otras enfermedades que pueden atacar a los seres humanos. El 12 de marzo de 2020, Will Douglas Heaven, de MIT News, explicó que se cree que la Inteligencia Artificial (IA) podría ayudar con la próxima pandemia, pero ya no con esta actual pandemia del Covid-19. Algunas cosas deben cambiar si queremos que la IA sea útil la próxima vez y, según Heaven, es posible que algunas no nos gusten. Fue un algoritmo de IA el que vio venir la pandemia por primera vez, o eso dice la historia. En efecto, el 30 de diciembre de 2019, una compañía de inteligencia artificial llamada BlueDot, que utiliza el aprendizaje automático para monitorear brotes de enfermedades infecciosas en todo el mundo, alertó a sus clientes, incluidos varios gobiernos, hospitales y empresas, sobre un aumento inusual en los casos de neumonía en Wuhan, China, que presagiaban alguna posible epidemia. Pero pasarían otros nueve días antes de que la Organización Mundial de la Salud (OMS) señalara oficialmente lo que todos conocemos como Covid-19. Ello demostró que la IA es capaz de detectar con gran rapidez y eficacia lo que los humanos tenemos mucha más dificultad de hacerlo.
Pero la empresa BlueDot no estaba sola. Un servicio automatizado llamado HealthMap, en el Boston Children’s Hospital, también detectó esos primeros signos. Al igual que un modelo dirigido por Metabiota, con sede en San Francisco, una empresa de análisis de riesgos que busca dificultar que las enfermedades infecciosas representen amenazas económicas para las empresas y los gobiernos. Pero que un algoritmo de IA pueda detectar un brote en el otro lado del mundo es bastante sorprendente, y ya sabemos que las advertencias tempranas salvan vidas. Pero, ¿cuánto ha ayudado realmente la IA para abordar el brote actual? Esta es una pregunta difícil de responder. Las empresas como BlueDot suelen tener dudas acerca de a quién proporcionan la información y de cómo se utiliza. Y algunos equipos científicos humanos afirman que detectaron el brote el mismo día que las IA. Otros proyectos en los que la IA se está explorando como una herramienta de diagnóstico o para ayudar a encontrar una vacuna todavía están en sus etapas iniciales. Incluso si tienen éxito, llevará tiempo, posiblemente meses, poner esas innovaciones en manos de los expertos de la salud que las necesiten. De hecho, la narrativa de que la IA es una nueva arma poderosa contra las enfermedades es solo en parte cierta y corre el riesgo de volverse contraproducente. Por ejemplo, demasiada confianza en las capacidades de AI podría conducir a decisiones poco informadas, que canalizasen dinero público a compañías de IA no probadas en lugar de intervenciones comprobadas, como programas en drogas farmacológicas. También las expectativas exageradas pero decepcionantes han provocado una disminución en el interés en la IA y la consiguiente pérdida de fondos. Así que aquí tenemos una comprobación de la realidad. Esta vez la IA no nos salvará del coronavirus. Pero hay muchas posibilidades de que juegue un papel más importante en futuras epidemias, si hacemos algunos cambios importantes. La mayoría de estos cambios no serán fáciles y, probablemente, no nos gustarán. Hay tres áreas principales en las que la IA podría ayudar: la predicción, el diagnóstico y el tratamiento. En el campo de la predicción tenemos empresas, como BlueDot y Metabiota, que utilizan una variedad de algoritmos de procesamiento en lenguaje natural para monitorizar los medios de comunicación e informes oficiales de atención médica, en diferentes idiomas y en todo el mundo. A partir de esta monitorización pueden señalar si mencionan enfermedades de alta prioridad, como el coronavirus u otras epidemias, como el VIH o la tuberculosis. Sus herramientas predictivas también pueden recurrir a datos de viajes aéreos para evaluar el riesgo de que los centros de tránsito puedan ver a personas infectadas llegando o saliendo de distintas zonas. Los resultados actuales son razonablemente precisos. Por ejemplo, un reciente informe público de Metabiota, el 25 de febrero de 2020, predijo que el 3 de marzo de 2020 habría 127.000 casos acumulativos en todo el mundo. Excedió en alrededor de 30.000, pero Mark Gallivan, director de ciencia de los datos de la empresa, dice que esto todavía está dentro del margen de error. También enumeró los países con mayor probabilidad de reportar nuevos casos, incluidos China, Italia, Irán y los Estados Unidos. De nuevo, acertaron bastante.

Otros algoritmos también vigilan las redes sociales. Stratifyd, una compañía de análisis de datos con sede en Charlotte, Carolina del Norte, está desarrollando una IA que escanea publicaciones en sitios como Facebook y Twitter y hace referencias cruzadas con descripciones de enfermedades tomadas de fuentes como los Institutos Nacionales de Salud, la Organización Mundial para Animal Health, y la base de datos global de identificadores microbianos, que almacena información de secuenciación del genoma. El trabajo de estas empresas muestra hasta qué punto el aprendizaje automático ha avanzado en los últimos años. Hace unos años, Google intentó predecir brotes con su desafortunado Flu Tracker, que se archivó en 2013 cuando no pudo predecir el pico de gripe de ese año. ¿Qué ha cambiado desde entonces? Se trata principalmente de la capacidad del último software para acceder una gama mucho más amplia de fuentes. El aprendizaje automático no supervisado también es clave. Permitir que una IA identifique sus propios patrones en la información que va detectando, en lugar de entrenarla en ejemplos preseleccionados, resalta hechos que quizás no se haya pensado buscar previamente. “Cuando haces predicciones, estás buscando un nuevo comportamiento“, dice el CEO de Stratifyd, Derek Wang. ¿Pero qué se hace con estas predicciones? La predicción inicial de BlueDot identificó correctamente un puñado de ciudades a las que se dirigía el virus. Esto podría haber permitido a las autoridades prepararse, alertar a los hospitales y establecer medidas de contención en una fase inicial. Pero a medida que crece el escalado de la epidemia, las predicciones se vuelven menos específicas. La advertencia de Metabiota de que ciertos países se verían afectados durante la semana siguiente podría haber sido correcta, pero es difícil saber qué hacer con esa información. Además, todos estos enfoques serán menos precisos a medida que progrese la epidemia, en gran parte porque los datos confiables del tipo de los que la IA necesita alimentarse han sido difíciles de obtener sobre Covid-19. Los noticiarios e informes oficiales ofrecen cuentas de infecciones y muertes bastante inconsistentes. También ha habido confusión sobre los síntomas y la forma en que el virus se transmite entre las personas. Los medios de comunicación pueden especular, mientras que las autoridades pueden minimizar el problema. Y predecir dónde se puede propagar una enfermedad desde cientos de lugares en docenas de países es una tarea mucho más desalentadora que avisar sobre dónde podría propagarse un brote en sus primeros días. “El ruido siempre es enemigo de los algoritmos de aprendizaje automático“, dice Derek Wang.
De hecho, Mark Gallivan reconoce que las predicciones diarias de Metabiota fueron más fáciles de hacer en las primeras dos semanas, más o menos. Uno de los mayores obstáculos es la falta de pruebas de diagnóstico, dice Gallivan. “Idealmente, tendríamos una prueba para detectar el nuevo coronavirus de inmediato y evaluar a todos al menos una vez al día“, dice. Tampoco sabemos realmente qué comportamientos están adoptando las personas, como quién trabaja desde casa, quién se pone en cuarentena, quién se lava las manos o no, o qué efecto podría tener. Si se desea predecir lo que sucederá a continuación, se necesita una imagen precisa de lo que está sucediendo en este momento. Tampoco está claro qué sucede dentro de los hospitales. Ahmer Inam, responsable de analítica avanzada en Pactera Edge, una consultora de datos e inteligencia artificial, dice que las herramientas de predicción serían mucho mejores si los datos de salud pública no estuvieran recluidos dentro de las agencias gubernamentales, como lo están en muchos países, incluido Estados Unidos. Esto significa que un algoritmo de IA debe apoyarse más en los datos fácilmente disponibles, como las noticias en línea. “Para cuando los medios de comunicación se den cuenta de una condición médica potencialmente nueva, ya es demasiado tarde“, dice Ahmer Inam. Pero si los algoritmos de IA necesita muchos más datos de fuentes confiables para ser útiles en esta área, las estrategias para obtenerla pueden ser controvertidas. Desgraciadamente para obtener mejores predicciones del aprendizaje automático, se necesitan compartir más datos personales con empresas y gobiernos, lo que implica menor privacidad de datos esenciales, como los médicos. Darren Schulte, CEO de Apixio, que ha creado una IA para extraer información de los registros de pacientes, cree que los registros médicos de todo Estados Unidos deberían abrirse para el análisis de datos. Esto podría permitir que una IA identifique automáticamente a las personas con mayor riesgo de Covid-19 debido a una afección subyacente. Los recursos podrían centrarse en aquellas personas que más los necesitan. La tecnología para leer registros de pacientes y extraer información que salva vidas existe, dice Schulte. El problema es que estos registros se dividen en múltiples bases de datos y son administrados por diferentes servicios de salud, lo que los hace más difíciles de analizar. “Me gustaría colocar mi IA en este gran océano de datos“, dice Schulte. “Pero nuestros datos se encuentran en pequeños lagos, no en un gran océano“.
Los datos de salud también deben compartirse entre los países, dice Ahmer Inam: “Los virus no operan dentro de los límites de las fronteras geopolíticas“. Cree que los países deberían verse obligados por acuerdo internacional a publicar datos en tiempo real sobre diagnósticos y admisiones hospitalarias, que luego podrían incorporarse a modelos del estudio de pandemia mediante aprendizaje automático a escala global. Por supuesto, esto puede ser complicado, ya que en diferentes partes del mundo tienen diferentes regulaciones de privacidad para los datos médicos. Y muchos de nosotros no queremos que nuestros datos médicos sean accesibles a terceros. Las nuevas técnicas de procesamiento de datos, como la privacidad diferencial y la capacitación sobre datos sintéticos en lugar de datos reales, podrían ofrecer una solución a este espinoso tema. La privacidad diferencial es un sistema para compartir públicamente información sobre un conjunto de datos al describir los patrones de los grupos dentro del conjunto de datos mientras se retiene información sobre las personas en el conjunto de datos. Otra forma de describir la privacidad diferencial es como una restricción en los algoritmos utilizados para publicar información agregada sobre una base de datos estadística que limita la divulgación de información privada de registros cuya información está en la base de datos. Por ejemplo, algunas agencias gubernamentales utilizan algoritmos diferencialmente privados para publicar información demográfica u otros agregados estadísticos al tiempo que garantizan la confidencialidad de las respuestas de las encuestas, y las compañías recopilan información sobre el comportamiento del usuario mientras controlan lo que es visible incluso para los analistas internos. Los datos sintéticos son la clave para hacer que el aprendizaje automático de la Inteligencia Artificial sea más rápido, y que se adopten algoritmos de la IA en nuestro día a día. Muchos expertos creen que los datos sintéticos son la clave para hacer que el aprendizaje automático de la Inteligencia Artificial sea más rápido y que se adopten algoritmos de la IA en nuestro día a día. Los datos sintéticos son datos artificiales fabricados por ordenadores, que los recopila de situaciones del mundo real. Estos datos son anónimos, y se crean con parámetros de cada usuario para que estos se asemejen al mundo real. Por ejemplo, se pueden crear datos sintéticos con los datos reales pero sin utilizar nombres, correos electrónicos, números de seguridad social o direcciones de esos datos. Los modelos generativos pueden aprender datos reales así como crear datos que sean parecidos a estos.
Estos datos sintéticos tienen muchas utilidades, entre ellas minimizar el tiempo, el coste y el riesgo de las operaciones. Los datos sintéticos tienen un gran potencial para las máquinas de aprendizaje profundo y los algoritmos de IA. Google, Amazon y Facebook tienen una ventaja considerable gracias a la cantidad de datos que crean a diario. Y es que la creación de datos sintéticos es más rentable que la recopilación de datos del mundo real la mayoría de las veces, y además algunas investigaciones evidencian que es posible obtener los mismos resultados utilizando datos sintéticos que datos de la realidad. El problema de los datos sintéticos es que es difícil crear datos de gran calidad. Si estos datos no son muy parecidos a los datos reales, se perderá calidad. Como son réplicas de propiedades específicas de un conjunto de datos reales, algunos comportamientos aleatorios pueden pasar desapercibidos. Los datos sintéticos pueden servir a nivel de registro para organizaciones médicas, para informar sobre los protocolos de atención y que la privacidad de los datos del paciente sea esencial. Pero esta tecnología aún se está perfeccionando. Encontrar un acuerdo sobre las normas internacionales llevará aún más tiempo. Por ahora, debemos aprovechar al máximo los datos que tenemos. La respuesta de Wang es asegurarse de que los humanos estén cerca para interpretar lo que los modelos de aprendizaje automático producen, asegurándose de descartar las predicciones que no suenan verdaderas. “Si uno es demasiado optimista o depende de un modelo predictivo totalmente autónomo, resultará problemático“, dice Wang. Las IA pueden encontrar señales ocultas en los datos, pero los humanos aún deben establecer relaciones. A fin de poder llevar a cabo un diagnostico temprano, además de predecir el curso de una epidemia, muchos esperan que la IA ayude a identificar a las personas infectadas. La IA tiene un historial se éxitos en este campo. Los modelos de aprendizaje automático para examinar imágenes médicas pueden detectar signos tempranos de enfermedades que los médicos humanos pasan por alto, desde enfermedades oculares hasta afecciones cardíacas o cáncer. Pero estos modelos generalmente requieren muchos datos para aprender. Se sugiere que el aprendizaje automático puede diagnosticar Covid-19 a partir de tomografías computarizadas de tejido pulmonar, si está capacitado para detectar signos reveladores de la enfermedad en las imágenes.
Alexander Selvikvåg Lundervold, de la Universidad de Ciencias Aplicadas de Noruega Occidental, en Bergen, Noruega, experto en aprendizaje automático e imágenes médicas, dice que debemos esperar que la IA pueda detectar eventualmente signos de Covid-19 en pacientes. Pero no está claro si la imagen es el camino a seguir. Por un lado, los signos físicos de la enfermedad pueden no aparecer en las exploraciones hasta algún tiempo después de la infección, por lo que no es muy útil como diagnóstico temprano. Además, dado que hasta el momento se dispone de muy pocos datos de capacitación, es difícil evaluar la precisión de los enfoques publicados en línea. La mayoría de los sistemas de reconocimiento de imágenes, incluidos los capacitados en imágenes médicas, están adaptados de los primeros modelos capacitados en ImageNet, un conjunto de datos ampliamente utilizado que abarca millones de imágenes cotidianas. “Para clasificar algo simple que está cerca de los datos de ImageNet, como las imágenes de perros y gatos, se puede hacer con muy poca información, pero hallazgos sutiles en imágenes médicas, no tanto“, dice Lundervold. Eso no quiere decir que no sucederá relativamente pronto y las herramientas de IA podrían construirse potencialmente para detectar las primeras etapas de la enfermedad en brotes futuros. Pero deberíamos ser escépticos sobre muchas de las afirmaciones de los médicos expertos en IA que diagnostican Covid-19 actualmente. Nuevamente, compartir más datos del paciente ayudará, y también lo harán las técnicas de aprendizaje automático que permiten entrenar modelos, incluso cuando hay pocos datos disponibles. Por ejemplo, el aprendizaje a partir de pocos datos, donde una IA puede aprender patrones de solo unos pocos resultados, y el aprendizaje de transferencia, donde una IA ya entrenada para hacer una cosa puede adaptarse rápidamente para hacer algo similar, son avances prometedores, pero aún está en fase experimental. Los datos también son esenciales para que la IA ayude a desarrollar tratamientos para la enfermedad. Una técnica para identificar posibles candidatos a fármacos es utilizar algoritmos de diseño generativo, que producen una gran cantidad de resultados potenciales y permite luego examinarlos para resaltar aquellos que vale la pena observar más de cerca.
Esta técnica se puede utilizar para buscar rápidamente a través de millones de estructuras biológicas o moleculares. Por ejemplo. SRI International está colaborando en el desarrollo de una herramienta de IA de este tipo, que utiliza el aprendizaje profundo para generar muchos nuevos fármacos candidatos que los científicos pueden evaluar para determinar su eficacia. Esto cambia las reglas del juego para el descubrimiento de drogas farmacológicas, pero aún pueden pasar muchos meses antes de que un candidato prometedor se convierta en un tratamiento viable. En teoría, las IA también podrían usarse para predecir la evolución del Covid-19. Ahmer Inam imagina ejecutar algoritmos de aprendizaje sin supervisión para simular todas las posibles rutas de evolución. Luego, podría agregar posibles vacunas a la mezcla y ver si los virus mutan para desarrollar resistencia. “Esto permitirá a los virólogos estar unos pasos por delante de los virus y crear vacunas en caso de que ocurra alguna de estas mutaciones del fin del mundo“, dice Inam. Es una posibilidad interesante, pero aún remota. Todavía no tenemos suficiente información sobre cómo muta el virus para poder simularlo. Mientras tanto, la barrera final pueden ser las autoridades políticas que gestionan una epidemia. “Lo que más me gustaría cambiar es la relación entre los encargados de formular políticas y la IA. La IA no podrá predecir brotes de enfermedades por sí misma, sin importar la cantidad de datos que obtenga. Hacer que los líderes del gobierno, las empresas y la atención médica confíen en estas herramientas cambiará fundamentalmente la rapidez con la que podemos reaccionar ante los brotes de enfermedades“, dice Wang. Pero esa confianza debe provenir de una visión realista de lo que la IA puede y no puede hacer ahora, así como lo que podría mejorar la próxima vez. Aprovechar al máximo la IA requerirá muchos datos, tiempo y coordinación inteligente entre muchas personas diferentes.
Pero antes de seguir con la IA intentemos saber algo más sobre este virus. Al igual que cuando se determinó, en los años ochenta del siglo XX, que el virus de la inmunodeficiencia humana VIH, que causa el sida, no había sido producido en ningún laboratorio, ahora un equipo multinacional de científicos llega a la conclusión de que el coronavirus SARS-CoV-2, que causa el Covid-19, tuvo su origen en la ciudad china de Wuhan, a finales del año pasado, producto de una evolución natural. En el análisis de los datos públicos de la secuencia del genoma del SARS-CoV-2 y los virus relacionados, los investigadores no encontraron evidencia de que el coronavirus se haya producido en un laboratorio o haya sido diseñado de otro modo, como se detalla en Nature Medicine. Ahora, como señala Kristian Andersen, del Centro de Investigación Biomédico Scripps Research y miembro del equipo que ha llevado a cabo el estudio, “al comparar los datos disponibles de la secuencia del genoma para las cepas conocidas de coronavirus, podemos determinar firmemente que el SARS-CoV-2 se originó a través de procesos naturales”. Además de Kristian Andersen, este equipo estuvo integrado por otros investigadores como Robert F. Garry, de la Universidad de Tulane, Edward Holmes, de la Universidad de Sydney, Andrew Rambaut, de la Universidad de Edimburgo, y W. Ian Lipkin, de la Universidad de Columbia. Los coronavirus son una gran familia de virus que pueden causar enfermedades que varían ampliamente en su nivel de severidad. La primera enfermedad grave conocida causada por un coronavirus surgió con la epidemia del Síndrome Respiratorio Agudo Severo (SRAS) de 2003, también en China. Un segundo brote de enfermedad grave comenzó en 2012, en Arabia Saudita, con el Síndrome Respiratorio del Medio Oriente (MERS). El 31 de diciembre del 2019, las autoridades chinas alertaron a la Organización Mundial de la Salud (OMS) del brote de una nueva cepa de coronavirus que causa una enfermedad grave y muy contagiosa, que posteriormente se denominó SARS-CoV-2. Poco después de que comenzara la epidemia, científicos chinos secuenciaron el genoma del SARS-CoV-2 y pusieron los datos a disposición de investigadores de todo el mundo. De todos modos, en cada país tienen que realizarse nuevas secuenciaciones para determinar posibles mutaciones del virus. Los datos de la secuencia genómica resultante han demostrado que las autoridades chinas detectaron rápidamente la epidemia y que el número de casos de COVID-19 ha aumentado debido a la transmisión de persona a persona después de una sola introducción en la población humana. Andersen y su equipo multinacional y multidisciplinar utilizaron estos datos de secuenciación para explorar los orígenes y la evolución del SARS-CoV-2, centrándose en varias características reveladoras del virus.
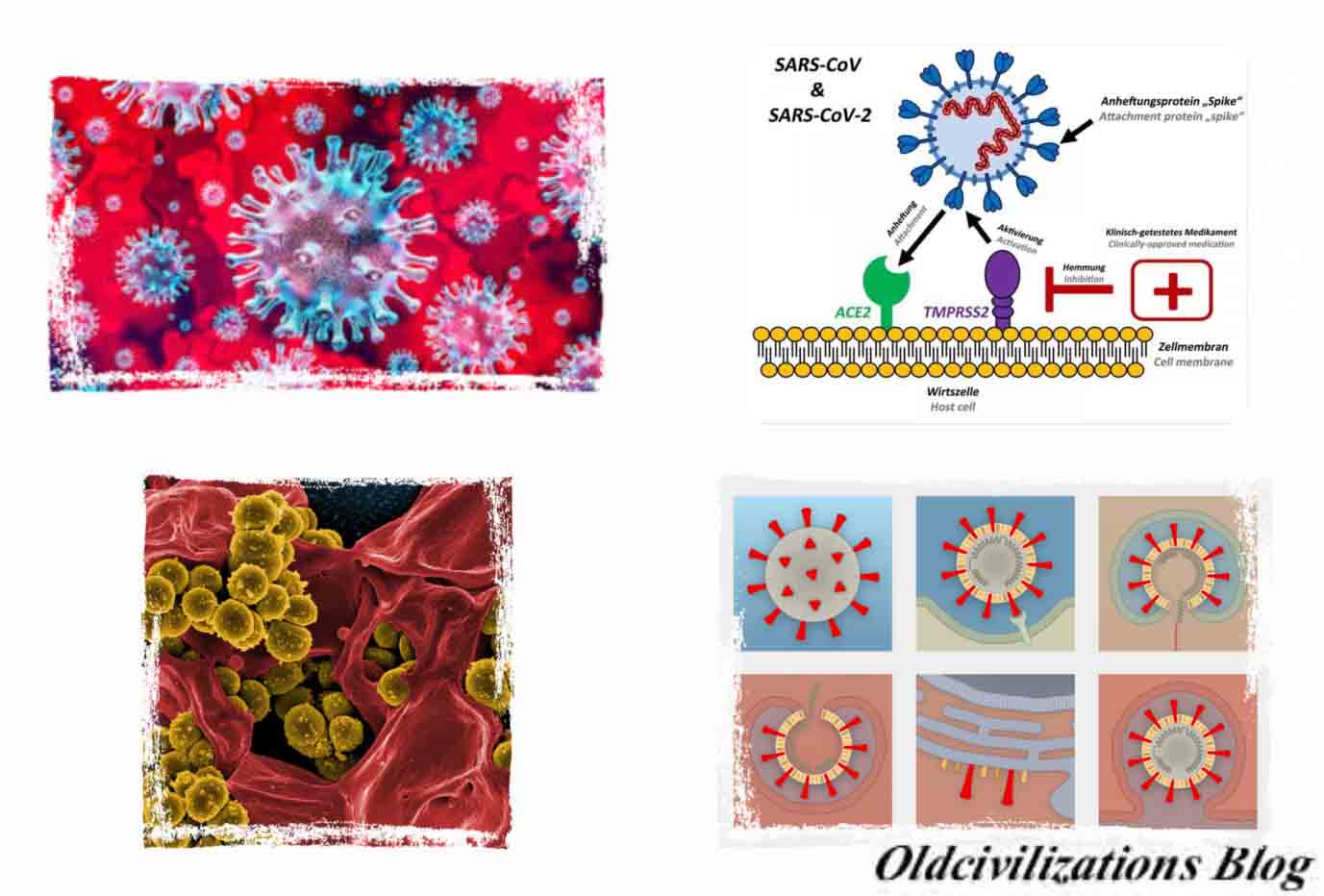
Así, los científicos analizaron la plantilla génica para las proteínas espiga, una especie de armaduras en el exterior del virus que utiliza para atrapar y penetrar las paredes externas de las células humanas y animales. Más específicamente, se centraron en dos características importantes de la proteína espiga: el dominio de unión al receptor (RBD), un tipo de gancho de agarre que se adhiere a las células huésped, y el sitio de escisión, un abridor de latas molecular que permite que el virus abra e ingrese en las células anfitrionas. Los científicos descubrieron que la porción RBD de las proteínas de la punta del SARS-CoV-2 había evolucionado para enfocarse en una característica molecular que hay en el exterior de las células humanas llamada ACE2 (enzima convertidora de angiotensina), un receptor involucrado en la regulación de la presión arterial. La proteína de la punta del SARS-CoV-2 es tan efectiva en la unión a las células humanas, de hecho, que los científicos concluyeron que era resultado de la selección natural y no producto de la ingeniería genética. Esta evidencia de evolución natural se respaldó por datos sobre la estructura molecular general del SARS-CoV-2. Si alguien buscara diseñar un nuevo coronavirus como patógeno, lo habrían construido a partir de la columna vertebral de un virus que se sabe que causa enfermedades. Pero los científicos descubrieron que el esqueleto del SARS-CoV-2 difería sustancialmente de los coronavirus ya conocidos y más bien se parecía a los virus relacionados que se pueden encontrar en los murciélagos y los pangolines. “Estas dos características del virus, las mutaciones en la porción RBD de la proteína espiga y su columna vertebral distinta, descartan la manipulación de laboratorio como un posible origen del SARS-CoV-2”, afirma Kristian Andersen. Por su parte, Josie Golding, responsable del departamento de Epidemiología de la Wellcome Trust de Londres, subraya que los hallazgos de Andersen y su equipo son “crucialmente importantes para aportar una visión basada en la evidencia de los rumores que han estado circulando sobre el origen no natural del virus (SARS-CoV-2) que causa Covid-19”. A partir de su análisis de secuenciación genómica, Andersen y su equipo analizaron los orígenes más probables para el SARS-CoV-2, siguiendo dos escenarios posibles. En el primero de los escenario, el virus habría evolucionado a su estado patógeno actual a través de la selección natural en un huésped no humano y luego habría saltado a las personas. Así es como habrían aparecido brotes previos de coronavirus en humanos al contraer el virus después de la exposición directa a civetas (SARS) y camellos (MERS).
Los investigadores propusieron a los murciélagos como el reservorio más probable para el SARS-CoV-2, ya que es muy similar a un coronavirus de murciélago. Sin embargo, no hay casos documentados de transmisión directa murciélago-persona, lo que sugiere que, probablemente, pudo haber un huésped intermedio entre murciélagos y humanos. En este escenario, las dos características distintivas de la proteína espiga del SARS-CoV-2, la porción RBD que se une a las células y el lugar de la escisión que abre el virus, habrían evolucionado a su estado actual antes de infectar a personas. En este caso, la epidemia actual probablemente surgiría rápidamente tan pronto como los humanos se infectaran, ya que el virus ya habría desarrollado las características que lo hacen patógeno y capaz de propagarse entre personas. En el otro escenario propuesto, una versión no patógena del virus habría saltado de un huésped animal a humanos y, luego, evolucionado a su estado patógeno actual dentro de la población humana. Por ejemplo, algunos coronavirus de pangolines, mamíferos tipo armadillo que se encuentran en Asia y África, tienen una estructura RBD muy similar a la del SARS-CoV-2. Un coronavirus de un pangolín podría haberse transmitido a un humano, ya sea directamente o a través de un huésped intermedio, como civetas o hurones. Entonces y como explican los investigadores, la otra característica de la proteína de espiga, el lugar de escisión, podría haber evolucionado dentro de un huésped humano, posiblemente a través de una circulación limitada no detectada en la población humana antes del comienzo de la epidemia.
Encontraron que el sitio de escisión del SARS-CoV-2 parece similar a los de cepas de gripe aviar que se ha demostrado que se transmite fácilmente entre personas. El SARS-CoV-2 podría haber desarrollado un lugar de escisión igual de virulento en células humanas y acelerar el inicio de la epidemia actual, ya que el coronavirus posiblemente se habría vuelto mucho más capaz de propagarse entre personas. El coautor del estudio, Andrew Rambaut, advierte que es difícil, si no imposible, saber en este momento cuál de los escenarios es más probable. Si el SARS-CoV-2 llegó a los humanos en su forma patógena actual de una fuente animal, aumenta la probabilidad de brotes futuros, ya que la cepa del virus que causa la enfermedad aún podría estar circulando entre la población animal y podría volver a saltar humanos. Las posibilidades de que un coronavirus no patógeno pase a la población humana y luego desarrolle propiedades similares al SARS-CoV-2 son menores. En cualquier caso, todo indica que el COVID-19 es producto de la evolución natural, con lo que se pone fin a cualquier especulación sobre su origen a través de ingeniería genética. La propagación de datos falsos y el desconocimiento de muchos, hace posible que esas afirmaciones calen en sociedad y, lo que es peor, se difundan. En 1994, a propósito del sida, un científico estadounidense de renombre aseguraba a quien le quería escuchar que el virus de la inmunodeficiencia humana no era el causante del sida. La situación se zanjó cuando el doctor Anthony Fauci, máximo responsable de los Institutos Nacionales de la Salud (INH) de EE UU, aceptó la invitación de una cadena de televisión para enfrentarse, dialécticamente hablando, con el autor del rumor. Con voz queda, el doctor Fauci, ante millones de espectadores, le dijo: “Si está tan seguro que el VIH no es el agente causante del sida, ¿por qué no se inocula usted mismo este retrovirus?”. El científico era Kary Mullis, recientemente fallecido, que consiguió el Nobel de Química por su hallazgo de la Reacción en Cadena de la Polimerasa (PCR).
Asimismo, antes de seguir con aplicaciones médicas de los algoritmos de IA, veamos algunos conceptos sobre Inteligencia Artificial. En los últimos años, los medios de comunicación han prestado una atención cada vez más grande a la inteligencia artificial (IA). El Aprendizaje Automático (Machine Learning) y el Aprendizaje Profundo (Deep Learning), ambos relacionados con la IA, han sido mencionados en numerosos artículos. Se nos promete un futuro de coches autónomos y asistentes digitales, un futuro algunas veces pintado con tintes pesimistas y otras veces de manera utópica, en donde el trabajo será escaso y la mayoría de la actividad económica será manejada por robots, computadoras y algoritmos de IA. Una pregunta que nos podemos hacer es si un computador con algoritmos de IA puede aprender por sí mismo como realizar una tarea específica, en vez de programadores especificando regla por regla como procesar los datos. ¿Podría un computador con algoritmos de IA aprender automáticamente estas reglas directamente de los datos a los que tiene acceso? A diferencia del clásico paradigma de la IA simbólica, donde seres humanos escriben las reglas, en forma de programa, y datos para ser procesados de acuerdo con dichas reglas, para así obtener respuestas o resultados de salida, con el Aprendizaje Automático los seres humanos pasamos los datos y las posibles respuestas, como ejemplos o casos, a partir de dichos datos como entrada, con el fin de obtener a la salida las reglas que nos permiten hacer el mapeo efectivo entre las entradas y sus correspondientes salidas. Estas reglas pueden ser luego aplicadas a nuevos datos para producir respuestas originales, es decir, generadas automáticamente por las reglas que el sistema “aprendió” y no por reglas explícitamente codificadas por programadores. Un sistema de Aprendizaje Automático es “entrenado” en vez de ser explícitamente “programado”. A este sistema se le presentan muchos ejemplos relevantes para la tarea en cuestión y este encuentra la estructura estadística o los patrones en dichos ejemplos, que eventualmente permiten al sistema aprender las reglas para automatizar dicha tarea. Por ejemplo, si deseamos automatizar la tarea del etiquetado de nuestras fotos de vacaciones, lo que haríamos es pasar al sistema de Aprendizaje Automático muchos ejemplos de fotos ya etiquetadas por humanos y el sistema aprendería las reglas estadísticas que le permitirían asociar fotos específicas con sus respectivas etiquetas.

Sin embargo, aunque el Aprendizaje Automático empezó a ser tenido en cuenta desde la década de1990, ahora se ha convertido en el más popular y exitoso de los sub-campos de la IA, una tendencia favorecida por la disponibilidad de mejor hardware y bases de datos gigantescas. El Aprendizaje Automático está fuertemente relacionado a la estadística matemática, pero con ciertas diferencias. A diferencia de la estadística, el Aprendizaje Automático tiende a lidiar con grandes y complejos conjuntos de datos, los cuales pueden contener millones de imágenes, cada una de ellas con miles de pixeles, para lo cual el clásico análisis estadístico, como el análisis bayesiano, sería inútil. Como resultado, el Aprendizaje Automático, y en especial el Aprendizaje Profundo, muestra poca teoría matemática, comparada con el campo de la estadística, y son considerados más como campos orientados hacia la ingeniería. Es decir, el Aprendizaje Automático es una disciplina aplicada, en la cual las ideas son probadas mucho más a menudo de forma empírica que de forma teórica. Para definir el Aprendizaje Profundo y entender la diferencia entre éste y otros enfoques del Aprendizaje Automático, primero necesitamos alguna idea de lo que hacen y cómo funcionan los algoritmos de Aprendizaje Automático. Acabamos de decir que el Aprendizaje Automático descubre reglas para ejecutar tareas de procesamiento de datos, dados los ejemplos de lo que se espera como respuesta o salida de dichos datos. Por lo tanto para realizar Aprendizaje Automático necesitaremos de tres ingredientes fundamentales: 1) Datos de entrada: Por ejemplo, si la tarea es reconocimiento de voz, estos datos de entrada serían archivos de sonido o grabaciones de gente hablando. Si la tarea es etiquetado de imágenes, estos datos podrían ser fotos o imágenes. 2) Ejemplos de lo que se espera como salida: En la tarea de reconocimiento de voz, estos podrían ser transcripciones generadas por humanos a partir de los archivos de audio. En la tarea de etiquetado de imágenes, las salidas esperadas pueden ser etiquetas tales como “oso”, “ratón”, “ser humano”, etc. 3) Una forma de medir si el algoritmo está realizando un buen trabajo: Este paso es necesario para determinar la distancia o desvío entre la salida actual generada por el algoritmo y la salida esperada. Esta medida es usada como señal de realimentación para ajustar la forma en la que el algoritmo trabaja y se actualiza. Este paso del ajustamiento es lo que llamamos propiamente “aprendizaje”. Estos elementos por si mismos son los fundamentales para todo tipo de algoritmo de Aprendizaje Automático y Aprendizaje Profundo. En el Aprendizaje Profundo, esta representaciones por capas son casi siempre aprendidas mediante modelos llamados Redes Neuronales, los cuales están literalmente estructurados en capas apiladas una después de la otra. El termino Redes Neuronales es una referencia a la neurobiología, pero, aunque algunos de los conceptos centrales en el Aprendizaje Profundo fueron desarrollados en parte de la inspiración tomada de nuestro entendimiento del cerebro, los modelos de Aprendizaje Profundo no son modelos del cerebro. No hay ninguna evidencia de que el cerebro implemente algunos de los mecanismos de aprendizaje usados en los modelos modernos de Aprendizaje Profundo.
A partir de lo indicado veremos ahora que es lo que realmente hace un algoritmo de Aprendizaje Automático y Aprendizaje Profundo para producir resultados que parecen salidos de la ciencia-ficción. Un modelo de Aprendizaje Automático transforma sus datos de entrada en respuestas con significado, un proceso que es “aprendido” de la exposición de dicho modelo a ejemplos previamente conocidos de entradas y salidas correspondientes. Por lo tanto, el problema central en el Aprendizaje Automático y el Aprendizaje Profundo es aprender representaciones útiles a partir de los datos de entrada, representaciones que nos acerquen a la generación o a la predicción de las salida esperadas. Pero, ¿qué es una representación? En su núcleo, una representación es una forma diferente de ver los datos, una forma diferente de representar o codificar los datos. Por ejemplo, una imagen a color puede ser codificada en formato RGB (red-green-blue) o en formato HSV (hue-saturation-value): Estas son dos representaciones diferentes de los mismos datos. Algunas tareas que pueden ser más difíciles utilizando una de esas representaciones pueden volverse mucho más fáciles al utilizar la otra representación. Por ejemplo, la tarea “seleccionar todos los pixeles rojos en una imagen” es mucho más simple en el formato RGB mientras que la tarea “hacer la imagen menos saturada” es más simple en el formato HSV. Los modelos de Aprendizaje Automático están diseñados para encontrar las representaciones más apropiadas a partir de la información que reciben como entrada, a partir de transformaciones de los datos que los hacen adecuados para la tarea en cuestión, así como en la tarea de clasificación de imágenes.
Y ahora entramos en el tema de la utilización de algoritmos de IA para combatir bacterias y virus, entre otros tipos de enfermedades. Según Steve Hanley, en un artículo del 22 de febrero de 2020, se está desarrollando un algoritmo de inteligencia artificial que crea antibióticos efectivos contra bacterias que se han vuelto resistentes a los antibióticos actuales. Pero esta experiencia puede servir para utilizar la IA para encontrar fármacos contra los virus, como el actual Covid-19, contra los que no sirven los actuales antibióticos. Sabemos que Alexander Fleming descubrió la penicilina por primera vez en 1928. Aunque su trabajo se basó en los esfuerzos de varios investigadores que lo precedieron, finalmente recibió el Premio Nobel de Medicina. En su discurso de aceptación, advirtió que las bacterias algún día podrían adaptarse a la penicilina y volverla menos útil, y eso es exactamente lo que sucedió. Hoy en día, hay muchas bacterias resistentes a los medicamentos que ignoran el efecto paliativo de la penicilina y muchos otros antibióticos que los médicos han recetado ampliamente, a menudo empujados por las compañías farmacéuticas. Crear nuevos medicamentos es un proceso complejo que puede ser extremadamente lento. Puede llevar años o a veces décadas desarrollar nuevos medicamentos, que es una de las razones por las que los precios de los medicamentos suelen ser extremadamente altos. Alguien tiene que pagar por todos los laboratorios y técnicos que llevan a cabo investigaciones sobre nuevos medicamentos año tras año. Muchas veces, los médicos ni siquiera saben si los medicamentos en los que están trabajando son efectivos hasta que están muy avanzados en su investigación. Los investigadores del MIT utilizaron un algoritmo de aprendizaje automático para identificar un medicamento basado en una molécula llamada HALicina, que mata muchas cepas de bacterias. La HALicina previno el desarrollo de resistencia a los antibióticos en la bacteria Escherichia coli (E.coli), una bacteria miembro de la familia de las enterobacterias y que forma parte de la microbiota del tracto gastrointestinal de animales homeotermos, como por ejemplo el ser humano. En cambio, la ciprofloxacina, un antibiótico del grupo de las fluoroquinolonas con efectos bactericidas, no lo hizo. Se vio que la inteligencia artificial puede ayudar a reducir drásticamente el tiempo que lleva descubrir nuevas drogas. Investigadores del MIT dicen que utilizando un algoritmo de IA han identificado un nuevo antibiótico que mata muchas bacterias resistentes a los medicamentos, según un informe de Science Daily. También resultó efectivo en dos ensayos con ratones infectados. Lo que hace que este descubrimiento sea aún más notable es que una computadora con este algoritmo de IA pudo realizar la tarea en solo tres días.
El algoritmo de IA está diseñado para identificar antibióticos potenciales que matan bacterias usando diferentes mecanismos y estrategias que los utilizados por las drogas existentes, según MIT News. Según James Collins, profesor de ingeniería médica y ciencia: “Queríamos desarrollar una plataforma que nos permitiera aprovechar el poder de la inteligencia artificial para marcar el comienzo de una nueva era de descubrimiento de antibiótico. Nuestro enfoque reveló esta increíble molécula que posiblemente sea uno de los antibióticos más potentes que se ha descubierto. Nos enfrentamos a una crisis creciente en torno a la resistencia a los antibióticos, y esta situación está siendo generada por un número cada vez mayor de agentes patógenos que se vuelven resistentes a los antibióticos existentes y una tubería anémica en las industrias biotecnológica y farmacéutica de nuevos antibióticos“. El algoritmo también identificó varios otros antibióticos potenciales que ahora se someterán a más pruebas. “El modelo de aprendizaje automático puede explorar grandes espacios químicos que pueden ser prohibitivamente costosos para los enfoques experimentales tradicionales“, dice Regina Barzilay, profesora de ingeniería eléctrica y ciencias de la computación en el MIT. Hasta ahora, el modelado por computadora era demasiado impreciso para producir resultados útiles, pero las redes neuronales más recientes pueden aprender automáticamente cómo lograr potenciales antibióticos utilizando la potencia de computadoras. Los investigadores diseñaron su nuevo modelo para buscar características químicas que hagan que las moléculas sean efectivas para matar la bacteria E.coli. Entrenaron el modelo con alrededor de 2.500 moléculas, incluidos alrededor de 1.700 medicamentos aprobados por la U.S. Food and Drug Administration (FDA) y un conjunto de 800 productos naturales con diversas estructuras y una amplia gama de actividades biológicas. Una vez que el modelo fue entrenado, los investigadores lo probaron en aproximadamente 6,000 compuestos. El modelo seleccionó una molécula que se predijo que tenía una fuerte actividad antibacteriana y tenía una estructura química diferente de cualquier antibiótico existente. Esa molécula se ha llamado HALicina, un nombre derivado de HAL, la computadora que aparece en la película de Stanley Kubrick: 2001 una odisea del espacio. Se cree que Kubrick eligió HAL porque todas las letras preceden a las iniciales IBM, que era el nombre de la compañía de computadoras dominante en el mundo en el momento de la película. Los investigadores lo probaron contra docenas de cepas bacterianas aisladas de pacientes y cultivadas en laboratorio y descubrieron que era capaz de matar a muchas que son resistentes al tratamiento antibiótico actual, incluidos Clostridium difficile, Acinetobacter baumannii y Mycobacterium tuberculosis.
El medicamento funcionó contra todas las especies de bacterias que probaron, con la excepción de Pseudomonas aeruginosa, un patógeno pulmonar difícil de tratar. MIT News añade que la HALicina puede matar bacterias al alterar su capacidad de producir ATP, una molécula que las células usan para almacenar energía. Los investigadores creen que las células tendrán dificultades para adaptarse a un proceso tan disruptivo. “Cuando se trata de una molécula que probablemente se asocia con componentes de membrana, una célula no puede adquirir necesariamente una sola mutación o un par de mutaciones para cambiar la química de la membrana externa. Mutaciones como esa tienden a ser mucho más complejas de adquirir evolutivamente”, dice Stokes. Después de identificar la HALicina, los investigadores utilizaron su modelo para examinar más de 100 millones de moléculas seleccionadas de la base de datos ZINC15, una colección en línea de aproximadamente 1.500 millones de compuestos químicos. La base de datos ZINC contiene una colección de compuestos químicos disponibles en el mercado preparados especialmente para la detección virtual . ZINC es utilizado por investigadores, como biólogos o químicos, por compañías farmacéuticas , compañías de biotecnología y universidades de investigación. Este examen, que tardó solo tres días, identificó 23 candidatos que eran estructuralmente diferentes de los antibióticos existentes y que se pronostica que no son tóxicos para las células humanas. En pruebas de laboratorio contra cinco especies de bacterias, los investigadores encontraron que ocho de las moléculas mostraban actividad antibacteriana, y dos eran particularmente poderosas. Los investigadores ahora planean probar estas moléculas más, y también para examinar más de la base de datos ZINC15. Los investigadores también planean usar su modelo para diseñar nuevos antibióticos y optimizar las moléculas existentes. Por ejemplo, podrían entrenar al modelo para agregar características que harían que un antibiótico en particular atacara solo ciertas bacterias, evitando que mate bacterias beneficiosas en el tracto digestivo de un paciente. Entonces, los científicos de hoy pueden hacer en tres días lo que normalmente lleva meses, años o incluso décadas para lograrlo. ¡Imagínense si ese tipo de poder de investigación puede aplicarse a la búsqueda de antídotos para todo tipo de virus y enfermedades!
Los antibióticos funcionan a través de una variedad de mecanismos, como el bloqueo de las enzimas involucradas en la biosíntesis de la pared celular, la reparación del ADN o la síntesis de proteínas. Pero la estrategia que sigue la HALicina no es convencional, ya que interrumpe el flujo de protones a través de la membrana celular. Los protones son partículas subatómicas con una carga eléctrica elemental positiva, que forma parte del núcleo del átomo, y cuya carga es igual en valor absoluto y de signo contrario a la del electrón, así como una masa 1836 veces superior a la de un electrón. En las pruebas iniciales con animales, la HALicina también parecía tener baja toxicidad y ser robusta contra la resistencia de las bacterias. Según Collins: “En los experimentos habituales, la resistencia a otros compuestos antibióticos generalmente surge dentro de un día o dos. Pero incluso después de 30 días de tales pruebas, no vimos ninguna resistencia contra la HALicina“. Luego, el algoritmo de IA examinó más de 107 millones de estructuras moleculares en una base de datos llamada ZINC15. De una lista de 23, las pruebas físicas identificaron 8 con actividad antibacteriana. Dos de estos tenían una potente actividad contra una amplia gama de patógenos y podían vencer incluso a cepas de E. coli resistentes a los antibióticos. El estudio es “un gran ejemplo del creciente cuerpo de trabajo que utiliza métodos computacionales para descubrir y predecir propiedades de posibles drogas“, dice Bob Murphy, biólogo computacional de la Universidad Carnegie Mellon en Pittsburgh. Señala que los métodos de IA se han desarrollado previamente para extraer grandes bases de datos de genes y metabolitos, cualquier molécula utilizada, capaz o producida durante el metabolismo, para identificar tipos de moléculas que podrían incluir nuevos antibióticos. Pero Collins y su equipo dicen que su enfoque es diferente: en lugar de buscar estructuras específicas o clases moleculares, están entrenando a su algoritmo de IA para buscar moléculas con una actividad particular. El equipo ahora espera asociarse con un grupo o compañía externa para incorporar la HALicina a los ensayos clínicos. También quiere ampliar el enfoque para encontrar más antibióticos nuevos y diseñar moléculas desde cero. Barzilay dice que su último trabajo es una prueba de concepto. “Este estudio lo reúne todo y demuestra lo que se puede hacer“.
Empezamos con una interesante entrevista que Kim Martineau, de MIT Quest for Intelligence, efectuó el 2 de abril de 2020 a Markus Buehler, músico y profesor del Instituto Tecnológico de Massachusetts (MIT). En esta entrevista se pregunta a Markus Buehler sobre la configuración del coronavirus SARS-CoV-2 (COVID-19) y las proteínas inspiradas en la IA para la música. Traducido al sonido, el SARS-CoV-2 (COVID-19) engaña a nuestro oído de la misma manera que el virus engaña a nuestras células. En la entrevista dice que las proteínas que componen todos los seres vivos son sensibles a la música. Pero, ¿qué son las proteínas? Las proteínas son moléculas formadas por aminoácidos que están unidos por un tipo de enlaces conocidos como enlaces peptídicos, un enlace químico que se establece entre el grupo carboxilo de un aminoácido y el grupo amino de otro aminoácido. El orden y la disposición de los aminoácidos dependen del código genético de cada persona. Todas las proteínas están compuestas por carbono, hidrógeno, oxígeno, nitrógeno, aunque la mayoría de proteínas contienen también azufre y fósforo. Las proteínas suponen aproximadamente la mitad del peso de los tejidos del organismo y están presentes en todas las células del cuerpo, además de intervenir en prácticamente todos los procesos biológicos que se producen. De entre todas las bio-moléculas, las proteínas desempeñan un papel fundamental en el organismo. Son esenciales para el crecimiento, gracias a su contenido de nitrógeno, que no está presente en otras moléculas como grasas o hidratos de carbono. También lo son para las síntesis y mantenimiento de diversos tejidos o componentes del cuerpo, como los jugos gástricos, la hemoglobina, las vitaminas, las hormonas y las enzimas, que actúan como catalizadores biológicos haciendo que aumente la velocidad a la que se producen las reacciones químicas del metabolismo. Asimismo, ayudan a transportar determinados gases a través de la sangre, como el oxígeno y el dióxido de carbono, y funcionan a modo de amortiguadores para mantener el equilibrio ácido-base y la presión oncótica del plasma, o presión osmótica coloidal, que es la presión osmótica debida a las grandes moléculas que se encuentran en disolución o suspensión coloidal. En el plasma sanguíneo la presión oncótica es ejercida principalmente por proteínas y complejos proteicos, sobre todo albúmina, y es una fuerza que tiende a empujar agua hacia el interior de los vasos sanguíneos o previene su salida, en oposición a la presión hidrostática que empuja el agua, y también solutos, hacia el exterior.
Otras funciones más específicas de las proteínas son, por ejemplo, las de los anticuerpos, un tipo de proteínas que actúan como defensa natural frente a posibles infecciones o agentes externos, además del colágeno, cuya función de resistencia lo hace imprescindible en los tejidos de sostén, y la miosina y la actina, dos proteínas musculares que hacen posible el movimiento, entre muchas otras. Las propiedades principales de las proteínas, que permiten su existencia y el correcto desempeño de sus funciones son la estabilidad y la solubilidad. La estabilidad hace referencia a que las proteínas deben ser estables en el medio en el que estén almacenadas o en el que desarrollan su función, de manera que su vida media sea lo más larga posible y no genere contratiempos en el organismo. En cuanto a la solubilidad, se refiere a que cada proteína tiene una temperatura y un pH que se deben mantener para que los enlaces sean estables. Las proteínas tienen también algunas otras propiedades secundarias, que dependen de las características químicas que poseen. Es el caso de la especificidad, en que su estructura hace que cada proteína desempeñe una función específica y concreta diferente de las demás y de la función que pueden tener otras moléculas. También la amortiguación de pH, ya que las proteínas pueden comportarse como ácidos o como bases, en función de si pierden o ganan electrones, y hacen que el pH de un tejido o compuesto del organismo se mantenga a los niveles adecuados. Asimismo, además tienen la capacidad electrolítica que les permite trasladarse de los polos positivos a los negativos y viceversa. Las proteínas son susceptibles de ser clasificadas en función de su forma y en función de su composición química. Según su forma, existen proteínas fibrosas, alargadas e insolubles en agua, como la queratina, el colágeno y la fibrina, así como las globulares, de forma esférica y compacta, y solubles en agua. Este es el caso de la mayoría de enzimas y anticuerpos, así como de ciertas hormonas. También existen las mixtas, con una parte fibrilar y otra parte globular. Dependiendo de la composición química que posean hay proteínas simples y proteínas conjugadas, también conocidas como heteroproteínas. Las simples se dividen a su vez en escleroproteínas y esferoproteínas. Las proteínas son esenciales en la dieta. Los aminoácidos que las forman pueden ser esenciales o no esenciales. En el caso de los primeros, no los puede producir el cuerpo por sí mismo, por lo que tienen que adquirirse a través de la alimentación. Son especialmente necesarias en personas que se encuentran en edad de crecimiento, como niños y adolescentes, y también en mujeres embarazadas, ya que hacen posible la producción de células nuevas. Están presentes sobre todo en los alimentos de origen animal como la carne, el pescado, los huevos y la leche. Pero también lo están en alimentos vegetales, como la soja, las legumbres y los cereales, aunque en menor proporción. Su ingesta aporta al organismo 4 kilocalorías por cada gramo de proteína.
Volviendo a Markus Buehler, vemos que desarrolla modelos de inteligencia artificial (IA) para diseñar nuevas proteínas, a veces traduciéndolas a un determinado sonido. Su objetivo es crear nuevos materiales biológicos para aplicaciones sostenibles y no tóxicas. En un proyecto con el MIT-IBM Watson AI Lab, Buehler está buscando una proteína para extender la vida útil de los alimentos perecederos. En un nuevo estudio en Extreme Mechanics Letters, él y sus colaboradores ofrecen un candidato prometedor. Se trata de una proteína de la seda hecha por abejas para su uso en la construcción de colmenas. En otro estudio reciente, en APL Bioengineering, fue un paso más allá y utilizó la IA para descubrir una proteína completamente nueva. Cuando ambos estudios se publcaron, el brote de Covid-19 estaba surgiendo en los Estados Unidos, y Buehler dirigió su atención a la proteína espiga del SARS-CoV-2, el apéndice que hace que el nuevo coronavirus sea tan contagioso. Él y sus colegas están tratando de obtener sus propiedades vibratorias a través de espectros de sonido basados en moléculas, que podrían ser clave para detener el virus. Pregunta: Su trabajo se centra en las proteínas hélice alfa que se encuentran en la piel y el cabello. ¿Por qué hace esta proteína tan intrigante? Debemos aclarar que las hélice alfa son estructuras secundarias de las proteínas. Esta hélice mantiene su forma por la presencia de los puentes de hidrógeno que se forman entre los átomos de oxígeno del grupo carbonilo de un aminoácido y el átomo de hidrógeno del grupo amino de otro aminoácido situado a cuatro aminoácidos de distancia en la cadena. En las proteínas, la hélice alfa es el principal motivo de estructura secundaria. Fue postulada primero por Linus Pauling, Robert Corey, y Herman Branson en 1951 basándose en las estructuras cristalográficas entonces conocidas de aminoácidos y péptidos. Respuesta: Las proteínas son los ladrillos y el mortero que forman nuestras células, órganos y cuerpo. Las proteínas hélice alfa son especialmente importantes. Su estructura tipo resorte les da elasticidad y resistencia, por lo que la piel, el cabello, las plumas, los cascos e incluso las membranas celulares son tan duraderas. Pero no solo son resistentes mecánicamente, sino que también tienen propiedades antimicrobianas incorporadas. Con IBM, estamos tratando de aprovechar este rasgo bioquímico para crear un recubrimiento de proteínas que pueda frenar el deterioro de los alimentos que se pudren rápidamente, como las fresas.
Pregunta: ¿Cómo escogieron la AI para producir esta proteína de seda? Respuesta: Capacitamos un modelo de aprendizaje profundo en el banco de datos de proteínas, que contiene las secuencias de aminoácidos y las formas tridimensionales de aproximadamente 120.000 proteínas. Luego alimentamos al modelo con un fragmento de una cadena de aminoácidos para la seda de abeja y le pedimos que predijera la forma de la proteína, átomo por átomo. Validamos nuestro trabajo sintetizando la proteína por primera vez en un laboratorio, un primer paso para desarrollar un recubrimiento antimicrobiano y estructuralmente duradero, que se pueda aplicar a los alimentos. Mi colega, Benedetto Marelli, se especializa en esta parte del proceso. También utilizamos la plataforma para predecir la estructura de las proteínas que aún no existen en la naturaleza. Así es como diseñamos nuestra proteína completamente nueva en el estudio APL Bioengineering. Pregunta: ¿Cómo mejora su modelo con otros métodos de predicción de proteínas? Respuesta: Utilizamos predicción de extremo a extremo. El modelo construye la estructura de la proteína directamente a partir de su secuencia, traduciendo patrones de aminoácidos en geometrías tridimensionales. Es como traducir un conjunto de instrucciones de IKEA en una estantería construida, menos la frustración. A través de este enfoque, el modelo aprende efectivamente cómo construir una proteína a partir de la proteína en sí, a través del lenguaje de sus aminoácidos. Sorprendentemente, nuestro método puede predecir con precisión la estructura de la proteína sin una plantilla. Supera a otros métodos de plegado y es significativamente más rápido que el modelado basado en la física. Debido a que el banco de datos de proteínas se limita a las proteínas que se encuentran en la naturaleza, necesitábamos una forma de visualizar nuevas estructuras para producir nuevas proteínas desde cero. Pregunta: ¿Cómo podría usarse el modelo para diseñar una proteína real? Respuesta: Podemos construir modelos átomo por átomo para secuencias encontradas en la naturaleza que aún no se han estudiado, como lo hicimos en el estudio de APL Bioingeniería, utilizando un método diferente. Podemos visualizar la estructura de la proteína y usar otros métodos computacionales para evaluar su función, analizando su estabilidad y las otras proteínas a las que se une en las células. Nuestro modelo podría usarse en el diseño de medicamentos o para interferir con las vías bioquímicas mediadas por proteínas en las enfermedades infecciosas.
Pregunta: ¿Cuál es el beneficio de traducir proteínas en sonido? Respuesta: Nuestros cerebros son excelentes para procesar sonidos. En un barrido, nuestros oídos captan todas sus características: tono, timbre, volumen, melodía, ritmo y acordes. Necesitaríamos un microscopio de alta potencia para ver un detalle equivalente en una imagen, y nunca podríamos verlo todo de una vez. El sonido es una forma muy elegante de acceder a la información almacenada en una proteína. Por lo general, el sonido se consigue haciendo vibrar un material, como una cuerda de guitarra, mientras que la música se hace al organizar los sonidos en determinados patrones. Con AI podemos combinar estos conceptos y usar vibraciones moleculares y redes neuronales para construir nuevas formas musicales. Hemos estado trabajando en métodos para convertir las estructuras de proteínas en representaciones audibles, y traducir estas representaciones en nuevos materiales. Pregunta: ¿Qué nos puede decir sobre convertir en sonidos la proteína espiga del SARS-CoV-2? Respuesta: Su pico de proteínas contiene tres cadenas de proteínas dobladas en un patrón misterioso. Estas estructuras son demasiado pequeñas para que el ojo humano las vea, pero se pueden escuchar. Representamos la estructura de la proteína física, con sus cadenas entrelazadas, como melodías entrelazadas que forman una composición en varias capas. Se pueden representar la secuencia de aminoácidos de la proteína espiga, sus patrones de estructura secundaria y sus intrincados pliegues tridimensionales. La pieza resultante es una forma de música de contrapunto, una técnica de improvisación y composición musical que evalúa la relación existente entre dos o más voces independientes (polifonía) con la finalidad de obtener cierto equilibrio armónico. Como una sinfonía, los patrones musicales reflejan la geometría de intersección de la proteína realizada al materializar su código ADN. Pregunta: ¿Qué aprendiste? Respuesta: El virus tiene una extraña habilidad para engañar y explotar al huésped para su propia multiplicación. Su genoma secuestra la maquinaria de fabricación de proteínas de la célula huésped y la obliga a replicar el genoma viral y producir proteínas virales para generar nuevos virus. Mientras se escucha, uno puede sorprenderse por el tono agradable e incluso relajante de la música. Pero engaña a nuestro oído de la misma manera que el virus engaña a nuestras células. Es un invasor disfrazado de visitante amigable. A través de la música, podemos ver el pico de SARS-CoV-2 desde un nuevo ángulo y apreciar la urgente necesidad de aprender el lenguaje de las proteínas.
Pregunta: ¿Puede abordarse con esta técnica el Covid-19 y el virus que lo causa? Respuesta: A largo plazo, sí. La traducción de proteínas en sonido les brinda a los científicos otra herramienta para comprender y diseñar proteínas. Incluso una pequeña mutación puede limitar o mejorar el poder patógeno del SARS-CoV-2. A través de la conversión en sonido también podemos comparar los procesos bioquímicos de su proteína espiga con coronavirus anteriores, como el SARS o el MERS. En la música que creamos, analizamos la estructura vibratoria de la proteína espiga que infecta al huésped. Comprender estos patrones vibratorios es fundamental para el diseño de medicamentos y otras acciones. Las vibraciones pueden cambiar a medida que las temperaturas aumentan, por ejemplo, y también pueden decirnos por qué la punta del SARS-CoV-2 gravita hacia las células humanas más que otros virus. Estamos explorando estas preguntas en investigaciones actuales y en curso con mis estudiantes graduados. También podríamos usar un enfoque compositivo para diseñar medicamentos para atacar el virus. Podríamos buscar una nueva proteína que coincida con la melodía y el ritmo de un anticuerpo capaz de unirse a la proteína espiga, lo que interfiere con su capacidad de infectar. Pregunta: ¿Cómo puede la música ayudar en el diseño de proteínas? Respuesta: Puedes pensar en la música como un reflejo algorítmico de la estructura. Las variaciones Goldberg, una composición musical para teclado, de Johann Sebastian Bach, por ejemplo, son una realización brillante del contrapunto, un principio que también hemos encontrado en las proteínas. Ahora podemos escuchar este concepto del contrapunto a medida que la naturaleza lo compuso en las proteínas, y compararlo con ideas en nuestra imaginación, o usar algoritmos de IA para hablar el lenguaje de diseño de las proteínas y dejar que imagine nuevas estructuras. Creemos que el análisis del sonido y la música puede ayudarnos a comprender mejor el mundo material. La expresión artística es, después de todo, solo un modelo del mundo dentro de nosotros y a nuestro alrededor.
Es importante aprender el lenguaje de las proteínas. Buehler explica que todo el concepto consiste en comprender mejor las proteínas y su amplia gama de variaciones. Las proteínas constituyen el material estructural de la piel, los huesos y los músculos, pero también son enzimas, señales químicas, interruptores moleculares y una gran cantidad de otros materiales funcionales que conforman la maquinaria de todos los seres vivos. Pero sus estructuras, incluida la forma en que se pliegan en las formas que a menudo determinan sus funciones, son extremadamente complicadas. Buehler explica que: “Tienen su propio idioma y no sabemos cómo funciona. No sabemos qué hace que una proteína de seda sea una proteína de seda o qué patrones reflejan las funciones que se encuentran en una enzima. No sabemos el código“. Al traducir ese lenguaje a una forma diferente a la que los humanos están particularmente en sintonía, y que permite que diferentes aspectos de la información se codifiquen en diferentes dimensiones, como tono, volumen y duración, Buehler y su equipo esperan obtener nuevas ideas sobre el relaciones y diferencias entre diferentes familias de proteínas y sus variaciones, y utilice estas ideas como una forma de explorar los muchos ajustes y modificaciones posibles de su estructura y función. Al igual que con la música, la estructura de las proteínas es jerárquica, con diferentes niveles de estructura en diferentes escalas de duración o tiempo. El nuevo método traduce una secuencia de aminoácidos de proteínas en una secuencia de sonidos de percusión y rítmicos. Luego, el equipo de Markus Buehler utilizó un sistema de inteligencia artificial para estudiar el catálogo de melodías producidas por una amplia variedad de proteínas diferentes. Hicieron que el sistema de IA introdujera ligeros cambios en la secuencia musical o creara secuencias completamente nuevas, y luego tradujera los sonidos nuevamente en proteínas que correspondían a las versiones modificadas o de nuevo diseño. Con este proceso, pudieron crear variaciones de las proteínas existentes como, por ejemplo, una que se encuentra en la seda de araña, uno de los materiales más fuertes de la naturaleza, haciendo así nuevas proteínas a diferencia de las producidas por la evolución natural. Los sonidos de percusión y rítmicos que se escuchan aquí se generan completamente a partir de secuencias de aminoácidos.
Aunque los propios investigadores pueden no conocer las reglas subyacentes, “la IA ha aprendido el lenguaje de cómo se diseñan las proteínas y puede codificarla para crear variaciones de versiones existentes o diseños de proteínas completamente nuevos“, dice Buehler. Dado que hay billones de combinaciones potenciales, cuando se trata de crear nuevas proteínas “no podrías hacerlo desde cero, pero eso es lo que la IA puede hacer“. Es un proceso de composición de nuevas proteínas. Al usar dicho sistema, entrenar el sistema de IA con un conjunto de datos para una clase particular de proteínas, puede llevar algunos días, pero luego puede producir en microsegundos un diseño para una nueva variante. Según Buehler: “Ningún otro método se acerca. La desventaja es que el modelo no nos dice qué está pasando realmente dentro. Simplemente sabemos que funciona“. Esta forma de codificar la estructura de proteínas en la música refleja una realidad más profunda. Buehler nos dice que: “Cuando miras una molécula en un libro de texto, es estática. Pero no es estático en absoluto. Se mueve y vibra. Cada parte de la materia es un conjunto de vibraciones. Y podemos usar este concepto como una forma de describir la materia“. El método aún no permite ningún tipo de modificaciones dirigidas a cualquier cambio en las propiedades, como la resistencia mecánica, la elasticidad o la reactividad química, sino que será esencialmente aleatorio. Buehler dice que: “Todavía tienes que hacer el experimento. Cuando se produce una nueva variante de proteína, no hay forma de predecir lo que hará“. El equipo también creó composiciones musicales desarrolladas a partir de los sonidos de aminoácidos, que definen esta nueva escala musical de 20 tonos. Las piezas de arte que construyeron consisten completamente en los sonidos generados a partir de aminoácidos. Buehler añade: “No se utilizan instrumentos sintéticos o naturales, lo que muestra cómo esta nueva fuente de sonidos se puede utilizar como una plataforma creativa“. A lo largo de los ejemplos se usan motivos musicales derivados de proteínas existentes de forma natural y proteínas generadas por IA, y todos los sonidos, incluidos algunos que se asemejan a bajos o tambores, también se generan a partir de los sonidos de aminoácidos. Los investigadores han creado una aplicación gratuita para teléfonos inteligentes Android, llamada Amino Acid Synthesizer, para reproducir los sonidos de los aminoácidos y grabar secuencias de proteínas como composiciones musicales.
Marc Meyers, profesor de ciencia de materiales en el Universidad de California en San Diego, que no participó en este trabajo, nos dice: “Markus Buehler ha sido dotado con el alma más creativa, y sus exploraciones en el funcionamiento interno de las biomoléculas están avanzando en nuestra comprensión de la respuesta mecánica de los materiales biológicos de la manera más significativa“. Meyers agrega: “El enfoque de esta imaginación en la música es una dirección novedosa e intrigante. Esta es la música experimental en su mejor momento. Los ritmos de la vida, incluidas las pulsaciones de nuestro corazón, fueron las fuentes iniciales de sonidos repetitivos que engendraron el maravilloso mundo de la música. Markus ha descendido al nanoespacio para extraer los ritmos de los aminoácidos, los componentes básicos de la vida“. “Las secuencias de proteínas son complejas, como lo son las comparaciones entre secuencias de proteínas“, dice Anthony Weiss, profesor de bioquímica y biotecnología molecular en la Universidad de Sydney, Australia, que tampoco estaba relacionado con este trabajo. El equipo del MIT “proporciona un enfoque impresionante, entretenido e inusual para acceder e interpretar esta complejidad. El enfoque se beneficia de nuestra capacidad innata para escuchar patrones musicales complejos. A través de la armonía ahora tenemos una herramienta entretenida y útil para comparar y contrastar secuencias de aminoácidos“. El equipo también incluyó al científico investigador Zhao Qin y Francisco Martin-Martinez en el MIT. El trabajo fue apoyado por la Oficina de Investigación Naval de los EE. UU. Y los Institutos Nacionales de Salud.
Es realmente sorprendente la idea de traducir proteínas a música y viceversa. Al convertir las estructuras moleculares en sonidos, los investigadores obtienen información sobre las estructuras de proteínas y crean nuevas variaciones. David L. Chandler, de la Oficina de noticias del MIT plantea la siguiente pregunta: ¿Quiere crear un nuevo tipo de proteína que pueda tener propiedades útiles? Y la respuesta es que no hay problema ya que, en un sorprendente matrimonio de ciencia y arte, los investigadores del MIT han desarrollado un sistema para convertir las estructuras moleculares de las proteínas, los componentes básicos de todos los seres vivos, en un sonido audible que se asemeja a pasajes musicales. Luego, invirtiendo el proceso, pueden introducir algunas variaciones en la música y convertirla nuevamente en nuevas proteínas nunca antes vistas en la naturaleza. Aunque no es tan simple como tararear la música de una nueva proteína, el nuevo sistema se acerca. Proporciona una forma sistemática de traducir la secuencia de aminoácidos de una proteína en una secuencia musical, utilizando las propiedades físicas de las moléculas para determinar los sonidos. Aunque los sonidos se transponen con el fin de ponerlos dentro del rango audible para los humanos, los tonos y sus relaciones se basan en las frecuencias vibratorias reales de cada molécula de aminoácidos, calculadas utilizando teorías de la química cuántica. Tal como ya hemos indicado, el sistema fue desarrollado por Markus Buehler, junto con el doctor Chi Hua Yu y otros. Como se describe en la revista ACS Nano, el sistema traduce los 20 tipos de aminoácidos, los bloques de construcción que se unen en cadenas para formar todas las proteínas, en una escala de 20 tonos. La secuencia larga de aminoácidos de cualquier proteína se convierte en una secuencia de notas. Si bien esta escala no suena familiar para las personas acostumbradas a las tradiciones musicales occidentales, los oyentes pueden reconocer fácilmente las relaciones y diferencias después de familiarizarse con los sonidos. Buehler dice que después de escuchar las melodías resultantes, ahora es capaz de distinguir ciertas secuencias de aminoácidos que corresponden a proteínas con funciones estructurales específicas.
Alguien podría preguntarse si puede democratizarse la inteligencia artificial en el cuidado de la salud. Una hackathon es un término usado en las comunidades hacker para referirse a un encuentro de programadores cuyo objetivo es el desarrollo colaborativo de software, aunque en ocasiones puede haber también un componente de hardware. Los hackathon promueven la colaboración entre médicos e ingenieros de software para facilitar el acceso ampliado a los registros médicos electrónicos, a fin de mejorar la atención al paciente. Kim Martineau, del MIT Quest for Intelligence, nos dice que un programa de inteligencia artificial es mejor que los médicos humanos para recomendar el tratamiento de una sepsis o septicemia, que es una afección médica grave, causada por una respuesta inmunitaria fulminante a una infección. El cuerpo libera sustancias químicas inmunitarias en la sangre para combatir la infección. Esta solución pronto puede ser susceptible de ensayos clínicos en Londres. El modelo de aprendizaje automático es parte de una nueva forma de practicar la medicina que extrae datos de registros médicos electrónicos para formas más efectivas de diagnosticar y tratar problemas médicos difíciles, incluida la sepsis, una infección de la sangre que mata a aproximadamente 6 millones de personas en todo el mundo cada año. El descubrimiento de una estrategia de tratamiento prometedora para la sepsis no se produjo de manera regular, a través de experimentos largos y cuidadosamente controlados. En cambio, surgió durante un hackathon en Londres en 2015. En una competencia que reunió a ingenieros y profesionales de la salud, un equipo encontró una mejor manera de tratar a los pacientes con sepsis en la unidad de cuidados intensivos, utilizando la base de datos MIMIC-III, una base de datos de cuidados críticos de libre acceso, del MIT. Un miembro del equipo, Matthieu Komorowski, trabajaría con los investigadores del MIT que supervisan el MIMIC-III para desarrollar un modelo de aprendizaje de refuerzo que predijera tasas de supervivencia más altas para los pacientes que recibieron dosis más bajas de líquidos IV y dosis más altas de drogas que constriñen los vasos sanguíneos. Casi todos los estados de shock circulatorio requieren una reposición de grandes volúmenes de líquidos IV, al igual que en la depleción grave de volumen intravascular, como la diarrea o el golpe de calor. La deficiencia de volumen intravascular se compensa en forma aguda por vasoconstricción, seguida luego de horas por una migración de líquidos desde el compartimiento extravascular al intravascular, lo que mantiene el volumen en la circulación a expensas del agua corporal total. Sin embargo, esta compensación se ve superada por las pérdidas mayores.
Los investigadores publicaron sus hallazgos en Nature Medicine. El documento es parte de un flujo de investigación que surgió de los datathon iniciados por Leo Celi, investigador del MIT y médico del personal del Centro Médico Beth Israel Deaconess. Un datathon es el punto de encuentro para todos los científicos de datos de todo el mundo que son desafiados con casos reales de la ciencia de los datos. Celi realizó el primer datathon en enero de 2014 para generar la colaboración entre enfermeras, médicos, farmacéuticos y científicos de datos del área de Boston. Cinco años después, un datathon ocurre una vez al mes en algún lugar del mundo. Después de meses de preparación, los participantes se reúnen en un hospital o universidad patrocinadora para pasar el fin de semana utilizando el MIMIC-III o una base de datos local en busca de mejores formas de diagnosticar y tratar a los pacientes de cuidados críticos. Muchos continúan publicando su trabajo, y en un nuevo hito para el programa, los autores del documento de aprendizaje de refuerzo ahora están preparando su modelo de tratamiento de sepsis para ensayos clínicos en dos hospitales afiliados al Imperial College de Londres. Como joven médico, Celi estaba preocupado por la amplia variación que vio en la atención al paciente. El tratamiento óptimo para el paciente promedio a menudo parecía inadecuado para los pacientes que encontró. En la década del 2000, Celi pudo ver cómo las nuevas y poderosas herramientas para analizar datos de registros médicos electrónicos podían personalizar la atención de los pacientes. Dejó su trabajo como médico para estudiar salud pública e informática biomédica en la Universidad de Harvard y el MIT, respectivamente. Al unirse al Instituto de Ingeniería y Ciencias Médicas del MIT después de graduarse, identificó dos barreras principales para una revolución en el uso de los datos en la atención médica: los profesionales médicos y los ingenieros rara vez interactuaban, y la mayoría de los hospitales, preocupados por la responsabilidad, querían conservar los datos de sus pacientes, dejando fuera del alcance de los investigadores todo, desde pruebas de laboratorio a las notas de los médicos. Celi pensó que un desafío estilo hackathon podría romper esas barreras. Los médicos aportarían una lluvia de ideas y las responderían con la ayuda de los científicos de datos y de la base de datos MIMIC-III. En el proceso, su trabajo demostraría a los administradores del hospital el valor de sus archivos sin explotar.
Finalmente, Celi esperaba que los hospitales de los países en desarrollo se inspiraran en ellos para crear también sus propias bases de datos. A los investigadores que no pudiesen pagar los ensayos clínicos se les podría ayudar para poder comprender mejor a sus propios pacientes y tratarlos mejor, democratizando la creación y validación de nuevos conocimientos. Celi afirma que: “La investigación no tiene que consistir en costosos ensayos clínicos. Una base de datos de registros de salud del paciente contiene los resultados de millones de mini experimentos que involucran a sus pacientes. De repente tienes varios cuadernos de laboratorio que puedes analizar y aprender”. Hasta ahora, varios hospitales patrocinadores, en Londres, Madrid, Tarragona, París, Sao Paulo y Beijing, se han embarcado en planes para construir su propia versión de MIMIC, que llevó a Roger Mark y Beth Israel del MIT siete años en su desarollo. Hoy el proceso es mucho más rápido gracias a las herramientas que el equipo de MIMIC-III ha desarrollado y compartido con otros para estandarizar y utilizar, de manera segura y manteniendo la privacidad, los datos de sus pacientes. Celi y su equipo se mantienen en contacto con sus colaboradores extranjeros mucho después de los datathons al hospedar investigadores en el MIT y reconectarse con ellos en datathons de todo el mundo. Según Celi: “Estamos creando redes regionales, en Europa, Asia y América del Sur, para que puedan ayudarse mutuamente. Es una forma de escalar y mantener el proyecto“. Humanitas Research Hospital, el más grande de Italia, está organizando el próximo Datathon de Critical Care de Milán, y Giovanni Angelotti y Pierandrea Morandini, estudiantes de intercambio recientes en el MIT, están ayudando a organizarlo. Marandini dice que: “La mayoría de las veces los médicos e ingenieros hablan diferentes idiomas, pero estos eventos promueven la interacción y generan confianza. No es como en una conferencia donde alguien está hablando y tú tomas notas. Tienes que construir un proyecto y llevarlo hasta el final. No hay experiencias como esta en el campo”. El ritmo de los eventos se ha acelerado con herramientas como Jupyter Notebook, Google Colab y GitHub, lo que permite a los equipos sumergirse en los datos al instante y colaborar durante meses después, acortando el tiempo de publicación. Celi y su equipo ahora enseñan un curso de un semestre en el MIT, Collaborative Data Science in Medicine, modelado a partir de los datos, creando una segunda vía para este tipo de investigación. Más allá de estandarizar la atención al paciente y hacer que la IA en el cuidado de la salud sea accesible para todos, Celi y sus colegas ven otro beneficio de los datos. Su proceso incorporado de revisión por pares podría evitar que se publiquen más investigaciones defectuosas. Describieron su caso en un artículo de 2016 en Science Translational Medicine. “Tendemos a celebrar la historia que se cuenta, no el código o los datos“, dice el coautor del estudio Tom Pollard, un investigador del MIT que forma parte del equipo MIMIC. “Pero el código y los datos son esenciales para evaluar si la historia es verdadera y la investigación es legítima“.
El 24 de marzo de 2020, Will Douglas Heaven, en MIT News, nos dice que se ha visto que una red neuronal de IA puede ayudar a detectar Covid-19 en las radiografías de tórax. COVID-Net podría ayudar a los científicos a desarrollar una herramienta de IA que pueda detectar signos reveladores. En efecto, una red neuronal de acceso abierto llamada COVID-Net, lanzada al público recientemente, podría ayudar a los investigadores de todo el mundo en un esfuerzo conjunto para desarrollar una herramienta de IA que pueda evaluar a las personas para detectar Covid-19. Pero, ¿qué es COVID-Net? Es una red neuronal convolucional, un tipo de red neuronal artificial donde las neuronas corresponden a campos receptivos de una manera muy similar a las neuronas en la corteza visual primaria de un cerebro biológico, y que. es un tipo de IA que es particularmente bueno para reconocer imágenes. Fue desarrollado por Linda Wang y Alexander Wong, de la Universidad de Waterloo, y la empresa de IA Darwin AI, en Canadá. COVID-Net fue entrenado para identificar signos de Covid-19 en radiografías de tórax utilizando 5.941 imágenes tomadas de 2.839 pacientes con diversas afecciones pulmonares, incluyendo infecciones bacterianas, infecciones virales que podían ser de Covid-19 o no. El conjunto de datos se proporciona junto con la herramienta para que los investigadores, o cualquier persona que quiera experimentar, puedan explorarlo y modificarlo. Además, varios equipos de investigación han anunciado herramientas de IA que pueden diagnosticar Covid-19 a partir de rayos X en las últimas semanas. Pero ninguno se ha puesto a disposición del público, lo que dificulta evaluar su precisión. La empresa Darwin AI está adoptando un enfoque diferente. Señala que COVID-Net no es aún una solución lista para producción y alienta a otros a ayudarlo a poderlo hacer. Darwin AI, cuyo CEO es Sheldon Fernández, también quiere que la herramienta explique su forma de razonar, lo que facilitará que los trabajadores de la salud lo utilicen. COVID-Net aún no se ha probado en un entorno real, pero sigue los pasos de una historia de éxito anterior. Muchos de los grandes avances en la visión por computadora en los últimos 10 años se deben al lanzamiento público de ImageNet, un gran conjunto de datos de millones de imágenes cotidianas, y AlexNet, una red neuronal convolucional que se capacitó en ella. Los investigadores han estado construyendo sobre ambos entornos desde entonces.
El 16 de marzo de 2020, Karen Hao, experta en Inteligencia Artificial del MIT Technology Review, escribió un artículo en que nos habla de que más de 24.000 trabajos de investigación sobre coronavirus están disponibles en un solo lugar. El conjunto de datos tiene como objetivo acelerar la investigación científica que podría combatir la pandemia de Covid-19. Los investigadores que colaboran en varias organizaciones lanzaron el conjunto de datos de investigación abierta Covid-19 (CORD-19), que incluye más de 24.000 artículos de investigación de revistas revisadas por pares, así como repositorios para ciencia biológica, como bioRxiv y medRxiv, donde los científicos pueden publicar sus trabajos. La investigación cubre el coronavirus SARS-CoV-2, así como Covid-19, el nombre científico de la enfermedad, así como otros grupos de coronavirus. Representa la colección más extensa de literatura científica relacionada con la pandemia en curso y continuará actualizándose en tiempo real a medida que se publiquen más investigaciones. La base de datos se compiló a petición de la Oficina de Política de Ciencia y Tecnología (OSTP) de la Casa Blanca, a través de una colaboración entre tres organizaciones: La Biblioteca Nacional de Medicina, Microsoft, investigación sin fines de lucro, y el Instituto Allen de Inteligencia Artificial. La Biblioteca Nacional de Medicina (NLM), a través de los Institutos Nacionales de Salud proporcionó acceso a publicaciones científicas existentes, y Microsoft usó sus algoritmos para literatura sanitaria para encontrar artículos relevantes. El Instituto Allen de Inteligencia Artificial (AI2) los convirtió de páginas web y archivos PDF en un formato estructurado que puede ser procesado por algoritmos de IA. La base de datos ahora está disponible en el sitio web Semantic Scholar de AI2. Como parte de su servicio mediante Semantic Scholar, que permite a la comunidad científica buscar fácilmente a través de la literatura académica, AI2 ya ha procesado el nuevo corpus utilizando las mismas técnicas de extracción y análisis de información que aplica a todas las investigaciones nuevas. Están emergiendo piezas clave de información, como autores, métodos, datos y citas, para facilitar a los científicos evaluar rápidamente cómo cada artículo se suma a la investigación existente.
También se están utilizando modelos de lenguaje natural de última generación para mapear las similitudes entre los documentos, como el modelo ELMo, una nueva técnica para incorporar palabras en el espacio vectorial real utilizando LSTM (Long Short Term Memory networks) bidireccionales capacitados en un objetivo de modelado de lenguaje, y el modelo BERT, reemplaza el modelado de lenguaje con un objetivo modificado que llamaron “modelado de lenguaje enmascarado“. En este modelo, las palabras en una oración se borran al azar y se reemplazan con una ficha especial (“enmascarada”) con una pequeña probabilidad, del 15%. Luego, se utiliza un Transformador para generar una predicción para la palabra enmascarada basada en las palabras sin máscara que la rodean, tanto a la izquierda como a la derecha. Este mapa ahora está impulsando una nueva característica en Semantic Scholar que permite a los investigadores crear una fuente de investigación personalizada basada en sus intereses. Es importante porque los científicos se apresuran contrarreloj para responder preguntas urgentes sobre la naturaleza del virus, con la esperanza de detener su propagación. La base de datos no solo les ayuda a consolidar la investigación existente en un solo lugar, sino que también hace que la literatura sea más fácil de extraer para obtener información, mediante algoritmos de procesamiento de lenguaje natural. La Oficina de Política de Ciencia y Tecnología (OSTP) ha lanzado una convocatoria abierta para que los investigadores de IA desarrollen nuevas técnicas para la minería de texto y datos que ayudarán a la comunidad médica a analizar la masa de información más rápidamente.
El 13 de marzo de 2020, Karen Hao, experta en Inteligencia Artificial del MIT Technology Review, dijo que los Centros para el Control y la Prevención de Enfermedades de Estados Unidos (CDC) están tratando de pronosticar la propagación del coronavirus. Han aprovechado uno de los mejores laboratorios de pronóstico de gripe del país para actualizar sus algoritmos de IA para la predicción de la pandemia de Covid-19 u otras que puedan surgir en el futuro. Cada año, los Centros para el Control y la Prevención de Enfermedades organizan una competencia para ver quién puede pronosticar con precisión la gripe. Los equipos de investigación en todo el país compiten con diferentes métodos, y los mejores resultados obtienen fondos y una asociación con la agencia para mejorar la preparación del país para la próxima temporada. Ahora la agencia está recurriendo a varias docenas de equipos para adaptar sus técnicas para pronosticar la propagación del coronavirus en un esfuerzo por tomar decisiones más informadas. Entre ellos hay un grupo en la Universidad Carnegie Mellon, en Pittsburgh, Pensilvania, que en los últimos años ha logrado algunos de los mejores resultados. El año pasado, el grupo fue designado como uno de los dos Centros Nacionales de Excelencia para Pronósticos de Gripe y se le pidió que dirigiera el diseño de un proceso de pronóstico a nivel comunitario. Roni Rosenfeld, jefe del grupo y del departamento de aprendizaje automático de la Universidad Carnegie Mellon, admite que inicialmente se mostró reacio a asumir las predicciones del coronavirus. Para una persona sin experiencia no parece que pronosticar las dos enfermedades, la gripe y el Covid-19, sea tan diferente, pero hacerlo para el nuevo brote es significativamente más difícil. A Rosenfeld le preocupaba si sus predicciones serían precisas y, por lo tanto, si serían útiles. Al final, estaba convencido de seguir adelante de todos modos. Según Rosenfeld: “Las personas actúan sobre la base de modelos de pronóstico, ya sea en papel o en la cabeza. Es mejor cuantificar estas estimaciones para poder discutirlas racionalmente en lugar de hacerlas en base a la intuición“. El laboratorio utiliza tres métodos para determinar el aumento y la disminución de los casos durante la temporada de gripe. El primero es lo que se conoce como un “pronóstico inmediato“: una predicción del número actual de personas infectadas. El laboratorio recopila datos recientes e históricos de los Centros para el Control y la Prevención de Enfermedades y otras organizaciones asociadas, incluidas las búsquedas de Google relacionadas con la gripe, la actividad de Twitter, el tráfico web en los Centros para el Control y la Prevención de Enfermedades, los sitios web médicos y Wikipedia. Esos flujos de datos se introducen en algoritmos de aprendizaje automático para hacer predicciones en tiempo real.
El segundo y el tercer método representan una predicción de lo que vendrá. Uno se basa en el aprendizaje automático y el otro en la opinión de servicios crowdsourcing, que sirve para reducir la carga de trabajo en el equipo de una empresa, buscando ideas nuevas e innovadoras en un sitio externo a la empresa, pero que podría tener algún tipo de relación con la empresa, por ejemplo, sus clientes. Las predicciones incluyen tendencias que se esperan hasta unas cuatro semanas, así como hitos importantes como cuándo llegará la temporada alta y el número máximo de casos esperados. Dicha información ayuda tanto a los Centros para el Control y la Prevención de Enfermedades como a los proveedores de atención médica, al aumentar la capacidad y prepararse con anticipación. El pronóstico de aprendizaje automático tiene en cuenta el pronóstico inmediato y los datos históricos adicionales de los Centros para el Control y la Prevención de Enfermedades. Hay 20 años de datos fiables sobre temporadas de gripe en los Estados Unidos, que proporcionan una amplia información para los algoritmos de IA. En cambio, el método de crowdsourcing aprovecha a un grupo de voluntarios. Cada semana, se les pide a expertos y no expertos que inicien sesión en un sistema en línea y revisen un cuadro que muestre la trayectoria de las temporadas de gripe pasadas y actuales. Luego se les pide que completen la curva de la temporada actual, proyectando cuántos casos de gripe más habrá con el tiempo. Aunque las personas no hacen muy buenas predicciones individualmente, en conjunto a menudo son tan buenas como el pronóstico de aprendizaje automático mediante IA. Con los años, el equipo de Roni Rosenfeld ha ajustado cada uno de sus métodos para predecir la trayectoria de la gripe con una precisión casi perfecta. Al final de cada temporada de gripe, los Centros para el Control y la Prevención de Enfermedades siempre actualizan retroactivamente los números finales, lo que le da al laboratorio de la Universidad Carnegie Mellon la oportunidad de ver cómo se acumulan sus proyecciones. Los investigadores ahora están adaptando todas las técnicas para el Covid-19, pero cada una planteará desafíos distintos. Para el pronóstico inmediato basado en el aprendizaje automático, muchas de las fuentes de datos serán las mismas, pero el modelo de predicción será diferente. Los algoritmos de IA necesitarán aprender nuevas correlaciones entre las señales en los datos y la realidad básica. Pero se ha producido un pánico mucho mayor en torno al coronavirus, que causa un patrón completamente diferente en la actividad en línea. Las personas buscarán información relacionada con el coronavirus en volúmenes mucho más altos, incluso si se sienten bien, lo que hace que sea más difícil saber quién puede tener síntomas.
En una situación de pandemia hay muy pocos datos históricos, lo que afectará los pronósticos. La gripe ocurre en un ciclo muy regular cada año, mientras que las pandemias son erráticas y poco frecuentes. La última pandemia, la gripe H1N1 en 2009, también tuvo características muy diferentes, afectando principalmente a las poblaciones más jóvenes que a las de edad avanzada. El brote de Covid-19 ha sido exactamente lo contrario, con pacientes mayores que se enfrentan a un mayor riesgo. Además de eso, los sistemas de vigilancia para el seguimiento de casos no estaban completamente desarrollados en ese momento. Según Rosenfeld: “Esa es la parte que creo que será la más difícil, porque los sistemas de aprendizaje automático, en su naturaleza, aprenden de los ejemplos“. Tiene la esperanza de que el método de crowdsourcing sea el más resistente. Por un lado, se sabe poco sobre cómo le irá en los pronósticos de pandemia. “Por otro lado, las personas son bastante buenas para adaptarse a circunstancias nuevas“. El equipo de Rosenfeld ahora está trabajando activamente en formas de hacer que estas predicciones funcionen lo mejor posible. Los laboratorios de pruebas de gripe ya están comenzando a hacer la transición a las pruebas de Covid-19 y a informar los resultados a los Centros para el Control y la Prevención de Enfermedades. El laboratorio de la Universidad Carnegie Mellon también se está acercando a otras organizaciones para obtener la mayor cantidad de datos lo más precisos posibles, como estadísticas anónimas y agregadas de registros de salud electrónicos y patrones de compra de medicamentos contra la fiebre, para encontrar señales más precisas, a fin de entrenar sus algoritmos de IA. Para compensar la falta de datos históricos de pandemias anteriores, el equipo confía en los datos más antiguos de la pandemia actual. Busca incorporar datos de países que fueron afectados anteriormente y actualizará sus modelos de aprendizaje automático a medida que se publiquen datos más precisos de forma retroactiva. Al final de cada semana, el laboratorio recibirá un informe de los Centros para el Control y la Prevención de Enfermedades con la trayectoria más actualizada de los casos en los Estados Unidos, incluidas las revisiones de las semanas anteriores. Luego, el laboratorio revisará sus modelos para cerrar las brechas entre las predicciones originales y las estadísticas continuas. Pero Rosenfeld se preocupa por las limitaciones de estos pronósticos. Hay mucha más incertidumbre que la que le permitiría sentirse cómodo. Por cada predicción que el laboratorio brinde a los Centros para el Control y la Prevención de Enfermedades, incluirá una gama de distintos porcentajes de posibilidades. Según Rosenfeld: “No vamos a decirte lo que va a pasar. Lo que te decimos es cuáles son las cosas que pueden suceder y qué tan probable es cada una de ellas“. Incluso después de que termine la pandemia, la incertidumbre no desaparecerá. Rosenfeld dice: “Será muy difícil saber qué tan buenos son nuestros métodos. Podrías ser exacto por las razones equivocadas. Podría ser inexacto por las razones equivocadas. Debido a que solo se tiene una temporada para probarlo, realmente no pueden sacarse conclusiones sólidas sobre su metodología”. Pero a pesar de todos estos desafíos, Rosenfeld cree que el trabajo valdrá la pena para informar a los Centros para el Control y la Prevención de Enfermedades y mejorar la preparación. Finaliza diciendo: “Puedo hacer lo mejor que puedo ahora. Es mejor que no tener nada“.
Se ha observado que el protocolo criptográfico permite una mayor colaboración en el descubrimiento de fármacos. La red neuronal de IA, que encuentra potenciales medicamentos de forma segura, podría potenciar la agrupación a gran escala de datos confidenciales. Rob Matheson, del MIT News, informó que los investigadores del MIT habían desarrollado un sistema criptográfico que podría ayudar a las redes neuronales a identificar candidatos prometedores a fármacos dentro de un conjunto de datos farmacológicos masivos, manteniendo la privacidad. El cálculo realizado a una escala tan grande podría permitir una amplia combinación de datos farmacológicos sensibles que facilitasen el descubrimiento predictivo de nuevos fármacos. Los conjuntos de datos de interacciones fármaco-objetivo (DTI), que muestran si los compuestos candidatos actúan sobre las proteínas objetivo, son fundamentales para ayudar a los investigadores a desarrollar nuevos medicamentos. Los modelos pueden ser entrenados para analizar conjuntos de datos de DTI conocidos y luego, utilizando esa información, encontrar nuevos candidatos a fármacos. En los últimos años, las empresas farmacéuticas, universidades y otras entidades se han abierto a agrupar datos farmacológicos en bases de datos más grandes que pueden mejorar en gran medida la capacitación de estos modelos de IA. Sin embargo, debido a cuestiones de propiedad intelectual y otras preocupaciones de privacidad, estos conjuntos de datos siguen siendo de alcance limitado. Los métodos de criptografía para proteger los datos tienen unas necesidades computacionales tan altas que no se adaptan bien a conjuntos de datos más allá de unas decenas de miles de DTI, que es una cantidad relativamente pequeña. En un artículo publicado en la revista Science, los investigadores del Laboratorio de Ciencias de la Computación e Inteligencia Artificial (CSAIL), del MIT, describen una red neuronal entrenada y probada de forma segura en un conjunto de datos de más de un millón de DTI. La red aprovecha las herramientas criptográficas modernas y las técnicas de optimización para mantener la privacidad de los datos de entrada, mientras se ejecuta de manera rápida y eficiente. Los experimentos del equipo muestran que la red funciona más rápido y con mayor precisión que los enfoques existentes, ya que puede procesar conjuntos de datos masivos en pocos días, mientras que otros marcos criptográficos tomarían meses. Además, la red identificó varias interacciones novedosas, incluida una entre el medicamento contra la leucemia Imatinib y una enzima ErbB4, cuyas mutaciones se han asociado con el cáncer, y que podrían tener importancia clínica.
Bonnie Berger, profesora de matemáticas de Simons en el MIT, especializada en ingeniería eléctrica y ciencias de la computación, además de ser responsable del grupo de computación y biología en el Laboratorio de Ciencias de la Computación e Inteligencia Artificial del MIT, nos dice que: “Las personas se dan cuenta de que necesitan agrupar sus datos para acelerar en gran medida el proceso de descubrimiento de fármacos y permitirnos, juntos, hacer avances científicos en la resolución de enfermedades humanas importantes, como el cáncer o la diabetes. Pero no tienen buenas formas de hacerlo. Con este trabajo, proporcionamos una forma para que estas entidades agrupen y analicen eficientemente sus datos a una escala muy grande“. Se han unido a Berger en el trabajo Brian Hie y Hyunghoon Cho, ambos estudiantes graduados en ingeniería eléctrica y ciencias de la computación e investigadores del grupo de Computación y Biología de CSAIL. El nuevo documento se basa en trabajos previos de los investigadores para proteger la confidencialidad del paciente en estudios genómicos, que encuentran vínculos entre variantes genéticas particulares y su incidencia en enfermedades. Esa información genómica podría revelar información personal, por lo que los pacientes pueden ser reacios a inscribirse en los estudios. En ese trabajo, Berger, Cho y un ex estudiante de doctorado de la Universidad de Stanford desarrollaron un protocolo basado en un marco de la criptografía llamado “intercambio secreto“, que analiza de manera segura y eficiente conjuntos de datos de un millón de genomas. En contraste, las propuestas existentes podrían manejar solo unos pocos miles de genomas. El intercambio secreto se utiliza en el cómputo multiparte, donde los datos confidenciales se dividen en “recursos compartidos” separados entre varios servidores. A lo largo del cómputo, cada parte siempre tendrá solo su parte de los datos, que parece completamente aleatoria. Sin embargo, colectivamente, los servidores aún pueden comunicarse y realizar operaciones útiles con los datos privados subyacentes. Al final del cálculo, cuando se necesita un resultado, las partes combinan sus acciones para revelar el resultado. “Utilizamos nuestro trabajo anterior como base para aplicar el intercambio secreto al problema de la colaboración farmacológica, pero no funcionó de inmediato“, dice Berger.
Una innovación clave fue reducir la computación necesaria durante la capacitación y las pruebas. Los modelos predictivos para el descubrimiento de fármacos representan las estructuras químicas y proteicas de los DTI (conjuntos de datos de interacciones fármaco-objetivo) como gráficos o matrices. Sin embargo, estos enfoques se escalan al cuadrado con el número de DTI en el conjunto de datos. Básicamente, el procesamiento de estas representaciones se vuelve computacionalmente extremadamente intenso a medida que crece el tamaño del conjunto de datos. “Si bien eso puede estar bien para trabajar con los datos sin procesar, si lo intenta en un cálculo seguro, no es factible“, dice Brian Hie. En cambio, los investigadores entrenaron una red neuronal que se basa en cálculos lineales, que se escalan de manera mucho más eficiente con los datos. “Necesitábamos una escalabilidad absoluta, porque estamos tratando de proporcionar una forma de agrupar datos en conjuntos de datos mucho más grandes“, dice Hyunghoon Cho. Los investigadores capacitaron a una red neuronal en la base de datos STITCH, que tiene 1,5 millones de DTI, lo que lo convierte en el mayor conjunto de datos disponible públicamente de este tipo. En el entrenamiento, la red codifica cada compuesto farmacológico y cada estructura proteica como una simple representación vectorial. Esto esencialmente condensa las estructuras complicadas como 1 y 0, que una computadora puede procesar fácilmente. A partir de esos vectores, la red aprende los patrones de interacciones y no interacciones. Alimentada con nuevos pares de compuestos y estructuras de proteínas, la red predice si interactuarán. La red también tiene una arquitectura optimizada para la eficiencia y la seguridad. Cada capa de una red neuronal requiere alguna función de activación que determina cómo enviar la información a la siguiente capa. En su red, los investigadores utilizaron una función de activación eficiente llamada unidad lineal rectificada (ReLU). Esta función requiere solo una comparación numérica segura y única de una interacción para determinar si se debe enviar (1) o (0) como datos a la siguiente capa, sin revelar nada sobre los datos reales. Esta operación puede ser más eficiente en computación segura en comparación con funciones más complejas, por lo que reduce la carga computacional al tiempo que garantiza la privacidad de los datos. “La razón por la que es importante es que queremos hacer esto dentro del marco de intercambio secreto y no queremos aumentar la sobrecarga computacional“, dice Berger. Al final, “no se revelan parámetros del modelo y todos los datos de entrada – los medicamentos, objetivos e interacciones – se mantienen privados”.
Los investigadores enfrentaron a su red contra varios modelos de texto sin sin cifrar de última generación en una parte de los DTI (conjuntos de datos de interacciones fármaco-objetivo) conocidos de DrugBank, una base de datos que contiene alrededor de 2000 DTI. Además de mantener los datos privados, la red de investigadores superó a todos los modelos en precisión de predicción. Solo dos modelos de línea de base podrían escalarse razonablemente a la base de datos STITCH, y el modelo de los investigadores logró casi el doble de precisión que esos modelos. Los investigadores también probaron pares de drogas-objetivo sin interacciones enumeradas en STITCH, y encontraron varias interacciones farmacológicas clínicamente establecidas que no estaban incluidas en la base de datos, pero que deberían estarlo. En el documento, los investigadores enumeran las principales predicciones más fuertes, que incluyen droloxifeno y un receptor de estrógenos, que alcanzaron ensayos clínicos de fase III como tratamiento para el cáncer de mama; y seocalcitol y un receptor de vitamina D para tratar otros tipos de cáncer. Cho y Hie validaron de forma independiente las interacciones recientes de mayor puntaje a través de organizaciones de investigación por contrato. El trabajo podría estar revolucionando el descubrimiento predictivo de drogas, dice Artemis Hatzigeorgiou, profesor de bio-informática en la Universidad de Tesalia, en Grecia. Según Hatzigeorgiou: “Habiendo entrado en la era de los grandes datos en fármaco-genética, es posible por primera vez recuperar un conjunto de datos de gran tamaño sin precedentes a partir de los datos de pacientes. Similar al procedimiento de aprendizaje de un cerebro humano, las redes neuronales artificiales necesitan una masa crítica de datos para proporcionar decisiones confiables. Ahora es posible el uso de millones de datos para entrenar una red neuronal artificial hacia la identificación de interacciones desconocidas entre drogas y objetivos. En tales condiciones, no es sorprendente que este modelo entrenado supere todos los métodos existentes en el descubrimiento de drogas farmacológicas“. A continuación, los investigadores están trabajando para establecer su canal de colaboración en un entorno del mundo real. “Estamos interesados en crear un entorno para la computación segura, para que podamos ejecutar nuestro protocolo seguro con datos reales“, dice Cho.
Stefanie Koperniak, del Departamento de Ingeniería Química del MIT, nos habla de la importancia del aprendizaje automático para los desafíos en la industria farmacéutica. Los investigadores y la industria relacionada con el MIT han formado un nuevo consorcio para ayudar al proceso de descubrimiento de fármacos. El MIT continúa sus esfuerzos para transformar el proceso de diseño y fabricación de medicamentos con un nuevo consorcio de la industria ligada al MIT, llamado Machine Learning for Pharmaceutical Discovery and Synthesis. El nuevo consorcio ya incluye ocho socios de la industria, prácticamente todas las principales empresas en el campo farmacéutico, como Amgen, BASF, Bayer, Lilly, Novartis, Pfizer, Sunovion y WuXi. Una gran cantidad de estas empresas farmacéuticas tienen sus centros de investigación en el área de Cambridge, en el estado de Massachusetts, Estados Unidos, cerca de la Universidad de Harvard y el Instituto Tecnológico de Massachusetts (MIT), lo que permite una estrecha cooperación y la creación de un centro para aplicaciones de inteligencia artificial (IA) en productos farmacéuticos. El proceso de descubrimiento de fármacos a menudo puede ser extremadamente costoso y lento, pero el aprendizaje automático ofrece enormes oportunidades para acceder y comprender, de manera más eficiente, a partir de grandes cantidades de datos químicos, con un gran potencial para mejorar tanto los procesos como los resultados. El consorcio tiene como objetivo romper la división entre la investigación del aprendizaje automático en el MIT y la investigación para el descubrimiento de fármacos, uniendo a los investigadores y la industria ligada al MIT, para identificar y abordar los problemas más importantes. Como parte de la iniciativa más amplia para unir el aprendizaje automático y la investigación de drogas farmacológicas, en abril el MIT organizó una cumbre dirigida por Regina Barzilay, profesora de informática de Delta Electronics, y Dina Katabi, profesora de ingeniería eléctrica e informática de Andrew, y Erna Viterbi, cofundadora de Qualcomm Andrew. La cumbre reunió a investigadores del MIT con líderes de tecnología, biotecnología y agencias reguladoras, para participar en encontrar fórmulas en que las tecnologías digitales y la inteligencia artificial puedan ayudar a abordar los principales desafíos en las industrias biomédica y de la atención médica.
Los primeros resultados para el consorcio comenzaron con el software y la tecnología financiados por el programa Make-It de la Agencia de Proyectos de Investigación Avanzada de Defensa (DARPA), que tiene el objetivo de integrar el aprendizaje automático con sistemas automatizados para la síntesis química. Los investigadores del MIT discutieron el potencial del consorcio con contactos en la industria farmacéutica, reuniéndose inicialmente con representantes de las compañías en mayo de 2017 y nuevamente en septiembre de 2017, momento en el que los investigadores de la industria y del MIT mostraron un gran interés en trabajar juntos. Desde entonces, a través del trabajo con la Oficina de Programas Patrocinados (OSP) del MIT y la Oficina de Licencias Tecnológicas del MIT (TLO), el consorcio se ha formado oficialmente. Una reunión del consorcio el 3 de mayo reunió a miembros de la industria e investigadores del MIT. “El entusiasmo de las empresas miembro y el potencial del aprendizaje automático crean una gran oportunidad para avanzar en la caja de herramientas para los científicos médicos en las industrias química y farmacéutica“, dice Klavs Jensen, profesor de ingeniería química y profesor de ciencias de los materiales e ingeniería de Warren K. Lewis. Según Jensen: “El aprendizaje automático puede ayudar a planificar las rutas de síntesis química y ayudar a identificar qué partes químicas dentro de una molécula contribuyen a propiedades particulares. Además, en última instancia, esto puede llevarnos a explorar nuevos espacios químicos, aumentar la diversidad química y darnos una mayor oportunidad para identificar compuestos adecuados que tendrán funciones biológicas específicas“. La reunión del 3 de mayo tuvo como objetivo presentar a los miembros de la industria los fundamentos del aprendizaje automático a través de tutoriales y proyectos de investigación conjuntos. Con este objetivo, Barzilay enseñó el primer tutorial sobre los conceptos básicos del aprendizaje supervisado. El tutorial cubrió modelos neuronales y se centró en el aprendizaje de la representación, con el objetivo de preparar a los participantes para presentaciones técnicas por la tarde. Barzilay nos dice que: “Estamos al comienzo de un campo relativamente inexplorado con infinitas oportunidades para una nueva ciencia, que tiene un impacto real en la vida de las personas. Nuestros colegas de la industria farmacéutica se preocupan por la ciencia de la manera en que lo hacemos en el MIT. Esta es la clave para una colaboración exitosa. Estoy continuamente aprendiendo de ellos y obteniendo nuevos problemas en los que pensar”.
Barzilay dice que uno de los objetivos del consorcio es establecer estándares de evaluación y crear conjuntos de datos de referencia para evaluar la precisión de los métodos de aprendizaje automático. Actualmente, la mayoría de los grupos de investigación evalúan sus resultados en base a conjuntos de datos patentados, lo que impide la comparación entre diferentes modelos y ralentiza el progreso científico. Para empeorar las cosas, muchos conjuntos de datos disponibles públicamente no son representativos de las complejidades reales que enfrentan los investigadores farmacéuticos. “Es para el beneficio de todos, tanto los investigadores como los usuarios de las nuevas tecnologías, comprender realmente dónde estamos y cuál es la verdadera capacidad de la nueva tecnología de aprendizaje automático“, dice Barzilay. Los investigadores principales del MIT para el consorcio abarcan diferentes áreas y departamentos, aportando experiencia en aprendizaje automático, química e ingeniería química. Además de los profesores Jensen y Barzilay, incluye a diversos investigadores. Según el investigador Tommi Jaakkola: “Al unir las ideas químicas con los conceptos y métodos modernos de aprendizaje automático, estamos abriendo nuevas vías para diseñar, comprender, optimizar y sintetizar drogas farmacológicas. El consorcio contribuye a elevar la química al ámbito de la ciencia de datos y a crear una nueva área interdisciplinaria similar a la biología computacional, con sus propias preguntas y objetivos clave. La colaboración también ofrece un nuevo campo de entrenamiento para estudiantes e investigadores por igual“. En la reciente reunión del consorcio, Connor Coley, un estudiante graduado en el Departamento de Ingeniería Química, que trabaja en los grupos de investigación de los profesores William H. Green y Klavs Jensen, presentó una descripción general y una demostración del software de planificación de síntesis, que podría tener un impacto especialmente significativo en el área de descubrimiento y desarrollo de moléculas pequeñas. Algunos aspectos de este trabajo de planificación de síntesis se resumen en el documento “Aprendizaje automático en la planificación de síntesis asistida por computadora“. Connor Coley dice que aunque el software de planificación de síntesis ha existido durante décadas, ningún sistema ha logrado una adopción generalizada.
Según Coley: “Ahora estamos en una posición única donde el acceso a grandes cantidades de datos químicos y el poder de cómputo ha permitido nuevos enfoques que finalmente podrían hacer que estas herramientas sean útiles y atractivas para los químicos en ejercicio. La planificación de síntesis tiene un papel claro en el descubrimiento en etapas tempranas, donde identificar rápidamente estrategias sintéticas para moléculas nuevas puede disminuir el tiempo de ciclo de las iteraciones de diseño-síntesis-prueba. Esperamos trabajar estrechamente con los miembros del consorcio para ayudar a facilitar el trabajo que realizan y ver nuestras metodologías y herramientas traducidas a la práctica”. Del mismo modo, los socios de la industria ven un gran valor y potencial en la implementación de enfoques de aprendizaje automático. “La aplicación de herramientas de aprendizaje automático brinda la oportunidad de aumentar y acelerar el descubrimiento y el desarrollo de medicamentos, y llevar nuevos medicamentos a los pacientes más rápidamente“, dice Shawn Walker, director de desarrollo de procesos de tecnologías fundamentales de sustancias farmacológicas en la farmacéutica Amgen. “Las herramientas de aprendizaje automático podrían ayudar a diseñar las mejores moléculas basadas en la afinidad de unión y minimizar la toxicidad, diseñar los mejores y más rentables procesos sintéticos para fabricar estas moléculas y extraer información de fuentes dispares, incluida la literatura química y las bases de datos de la compañía. Las posibilidades son infinitas, y esperamos que asociar el mejor talento científico con las mejores herramientas de aprendizaje automático conducirá a mejores resultados para los pacientes“. “Estamos entusiasmados de participar en este consorcio de aprendizaje automático del MIT, junto con nuestros otros socios de la industria“, dice José Duca, jefe de descubrimiento de fármacos, asistidos por computadora, en los Institutos Novartis para la Investigación Biomédica. “Este consorcio abordará el desafío de una planificación de rutas eficiente y focalizada, utilizando enfoques de aprendizaje automático de última generación. En última instancia, esperamos que esto acelere nuestra capacidad de fabricar medicamentos más seguros y potentes contra las enfermedades humanas“.
Guiada por la Inteligencia Artificial, una plataforma robótica automatiza la fabricación de moléculas. El nuevo sistema podría liberar a los químicos de tareas que consumen mucho tiempo, además de poder ayudar a descubrir nuevas moléculas. Becky Ham, corresponsal de MIT News, nos explica que un sistema desarrollado por investigadores del MIT está a un paso de automatizar la producción de pequeñas moléculas, que podrían usarse en medicina, energía solar y química de polímeros. El sistema, descrito en la revista Science, podría liberar a los químicos de una variedad de tareas rutinarias y que consumen mucho tiempo, así como puede ayudar a sugerir posibilidades sobre cómo hacer nuevos compuestos moleculares, según los líderes del estudio, Klavs F. Jensen, Connor W. Coley, el profesor Warren K. Lewis, de Ingeniería Química, Timothy F. Jamison, así como el profesor Robert R. Taylor, de Química y rector asociado en el MIT. Esta tecnología “promete ayudar a las personas a eliminar todas las partes tediosas de la construcción de moléculas, incluida la búsqueda de posibles vías de reacción y la construcción de los componentes de una línea de ensamblaje molecular, cada vez que se produce una nueva molécula“, dice Jensen. “Y como químico, puede darte inspiración para nuevas reacciones en las que no habías pensado antes“, agrega. El nuevo sistema combina tres pasos principales. Primero, el software, guiado por inteligencia artificial, sugiere una ruta para sintetizar una molécula y luego los químicos expertos revisan esta ruta y la refinan en una “receta” química. Finalmente la receta se envía a una plataforma robótica que ensambla automáticamente el hardware y realiza las reacciones que construyen la molécula. Connor W. Coley y sus colegas han estado trabajando durante más de tres años para desarrollar el paquete de software de código abierto que sugiere y prioriza posibles rutas de síntesis. En el corazón del software se encuentran varios modelos de redes neuronales, que los investigadores capacitaron en millones de reacciones químicas publicadas previamente y extraídas de las bases de datos Reaxys y de la Oficina de Patentes y Marcas de Estados Unidos. Reaxys, de Elsevier, es una herramienta basada en la web para la recuperación de información química y datos de la literatura publicada, incluyendo revistas y patentes. La información incluye compuestos químicos, reacciones químicas, propiedades químicas, datos bibliográficos relacionados, datos de sustancias con información de planificación de síntesis, así como procedimientos experimentales de revistas y patentes seleccionadas.
El software utiliza estos datos para identificar las transformaciones de reacción y las condiciones que cree que serán adecuadas para construir un nuevo compuesto. “Ayuda a tomar decisiones de alto nivel sobre qué tipos de intermedios y materiales de partida usar, y luego efectuar un análisis un poco más detallado sobre qué condiciones es posible que desee usar y si esas reacciones pueden ser exitosas“, dice Coley. “Una de las principales motivaciones detrás del diseño del software es que no solo te da sugerencias de moléculas que conocemos o reacciones que conocemos. Se puede generalizar a nuevas moléculas que nunca se han hecho“. añade Coley. Luego, los químicos revisan las rutas de síntesis sugeridas producidas por el software a fin de construir una receta más completa para la molécula objetivo. Los químicos a veces necesitan realizar experimentos de laboratorio o retocar con concentraciones de reactivos y temperaturas de reacción, entre otros cambios. “Toman parte de la inspiración de la IA y la convierten en un archivo de receta ejecutable, en gran parte porque la literatura química en este momento no tiene suficiente información para pasar directamente de la inspiración a la ejecución en un sistema automatizado“, dice Jamison. La receta final se carga en una plataforma donde un brazo robótico ensambla reactores modulares, separadores y otras unidades de procesamiento en una ruta de flujo continuo, conectando bombas y líneas que incorporan los ingredientes moleculares. “Carga la receta, eso es lo que controla la plataforma robótica, carga los reactivos y presiona iniciar, y eso le permite generar la molécula de interés“, dice el investigador Dale A. Thomas III. “Y luego, cuando se completa, vacía el sistema y puede cargar el siguiente conjunto de reactivos y recetas, y dejar que se ejecute“. A diferencia del sistema de flujo continuo que los investigadores presentaron anteriormente, que tuvo que configurarse manualmente después de cada síntesis, el nuevo sistema está completamente configurado por la plataforma robótica. “Esto nos da la capacidad de secuenciar una molécula tras otra, así como generar una biblioteca de moléculas en el sistema, de forma autónoma“, dice Jensen. El diseño de la plataforma, que tiene un tamaño de aproximadamente dos metros cúbicos, un poco más pequeño que una campana de extracción química estándar, se asemeja a una centralita telefónica y un sistema de operador que mueve las conexiones entre los módulos en la plataforma.
“El brazo robótico es lo que nos permitió manipular las rutas fluídicas, lo que redujo el número de módulos de proceso y la complejidad fluídica del sistema, y al reducir la complejidad fluídica podemos aumentar la complejidad molecular“, dice Dale A. Thomas III. “Eso nos permitió agregar pasos de reacción adicionales y expandir el conjunto de reacciones que podrían completarse en el sistema dentro de una huella relativamente pequeña“. Los investigadores probaron el sistema completo creando 15 pequeñas moléculas medicinales diferentes con diferente complejidad de síntesis, mediante procesos que toman entre dos horas para las creaciones más simples y aproximadamente 68 horas para fabricar múltiples compuestos. El equipo sintetizó una cierta variedad de compuestos, tales como aspirina, el antibiótico secnidazol en procesos consecutivos, el analgésico lidocaína y el medicamento contra la ansiedad diazepam en procesos consecutivos, utilizando una materia prima común de reactivos, así como el anticoagulante warfarina y el medicamento contra la enfermedad de Parkinson, safinamida, a fin de poder mostrar cómo el software podría diseñar compuestos con componentes moleculares similares pero con estructuras tridimensionales diferentes. También se sintetizaron una familia de cinco fármacos inhibidores de ACE y una familia de cuatro no esteroideos, fármacos anti-inflamatorios. “Estoy particularmente orgulloso de la diversidad de la química y los tipos de diferentes reacciones químicas“, dice Jamison, quien dijo que el sistema manejó unas 30 reacciones diferentes en comparación con aproximadamente 12 reacciones diferentes en el sistema de flujo continuo anterior. “Realmente estamos tratando de cerrar la brecha entre la generación de ideas de estos programas y lo que se necesita para ejecutar una síntesis“, dice Coley. “Esperamos que los sistemas de próxima generación aumenten aún más la fracción de tiempo y esfuerzo para que los científicos puedan enfocar sus esfuerzos en la creatividad y el diseño“. La investigación fue apoyada, en parte, por el programa Make-It de la Agencia de Proyectos de Investigación Avanzada de Defensa de los Estados Unidos (DARPA). El programa Make-It tiene como objetivo desarrollar un sintetizador químico automatizado para un descubrimiento y producción más rápidos de nuevas moléculas sintéticas. Aunque siendo un organismo militar no sabemos qué otras intenciones puede tener Make-It.
Las moléculas sintéticas son la base de muchos productos críticos para la misión del Departamento de Defensa de Estados Unidos, desde ingredientes farmacéuticos activos que se encuentran en un botiquín hasta materiales en baterías modernas y celdas de combustible. Sin embargo, los procesos actuales para diseñar y producir nuevas moléculas sintéticas son muy lentos y pueden tomar años entre el diseño inicial de una solución molecular y cuando está disponible para su uso en grandes cantidades. El programa Make-It de DARPA busca superar este desafío mediante el desarrollo de un sintetizador automatizado que pueda transformar materias primas simples en moléculas conocidas o nuevas definidas por el usuario. El objetivo de Make-It es desarrollar un sintetizador químico totalmente automatizado que pueda producir, purificar, caracterizar y escalar una amplia gama de moléculas pequeñas. Acelerar la tasa de descubrimiento y producción de moléculas podría acelerar los avances en una serie de áreas importantes para la seguridad nacional. “La síntesis es un cuello de botella para el descubrimiento y la producción de nuevas moléculas“, dijo Tyler McQuade, gerente de programa en la Oficina de Ciencias de Defensa de DARPA. “Los sintetizadores automatizados existen comercialmente para sistemas discretos como los biopolímeros naturales, pero la tecnología no es aplicable en todas las moléculas y la ampliación es difícil. La visión de Make-It es crear un sintetizador automatizado que produzca una amplia variedad de moléculas pequeñas complejas a escala de producción en semanas en lugar de años “. Si tiene éxito, Make-It ampliaría el acceso a la química sintética más allá de los químicos, permitiendo a los no químicos de otras disciplinas científicas aprovechar el poder de síntesis para nuevas aplicaciones. Otro beneficio sería revolucionar la reproducibilidad. Actualmente, la transferencia de conocimiento es un desafío en la química sintética, porque las condiciones y las configuraciones de reacción varían entre las ubicaciones de los laboratorios en todo el mundo. Con un sintetizador Make-It, las rutas químicas para nuevas moléculas podrían ser fácilmente replicadas por otros tan solo como enviando un mensaje de texto. Además, Make-It haría la síntesis más segura y ecológica al permitir un mejor control de las entradas del proceso, como los materiales de partida y los solventes, así como las condiciones como la temperatura y la presión. “Un sistema que pueda automatizar el diseño y las pruebas de rutas químicas específicas, haciendo un seguimiento tanto del éxito como de los fracasos, marcará el comienzo de la próxima fase del descubrimiento sintético“, dijo McQuade. El programa Make It busca experiencia en áreas tales como matemáticas, química computacional, ingeniería química, química analítica, ingeniería / control de procesos, química orgánica sintética, formulación y fabricación.
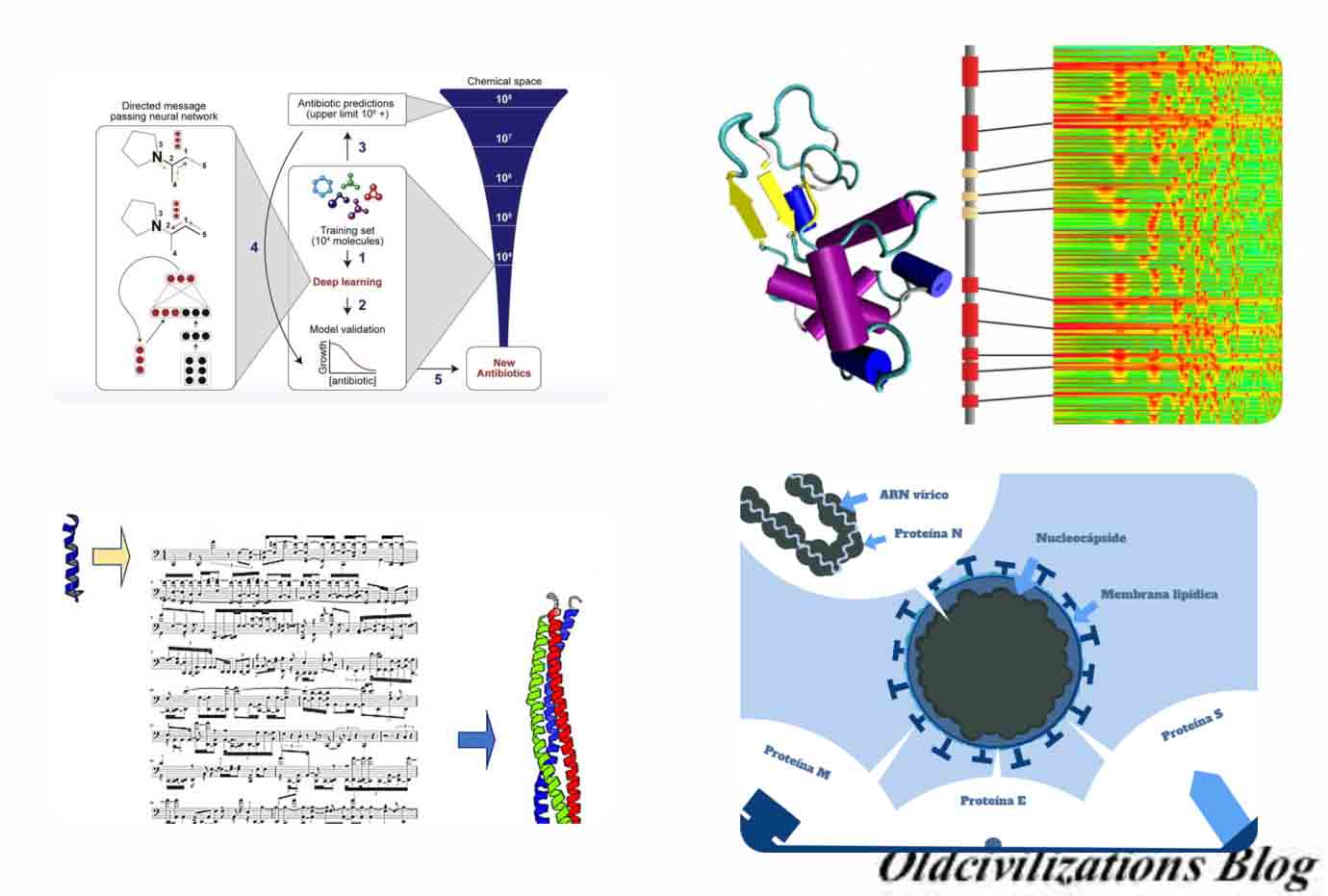
Se desarrolla un modelo que mejora la predicción del riesgo de mortalidad en pacientes de la UCI. Al capacitar a pacientes agrupados por estado de salud, la red neuronal puede estimar mejor si los pacientes morirán en el hospital o no. Rob Matheson, de MIT News, nos explica que en las unidades de cuidados intensivos, donde los pacientes entran con una amplia gama de condiciones de salud, el tratamiento depende en gran medida del juicio clínico. El personal de la UCI realiza numerosas pruebas fisiológicas, como análisis de sangre y control de signos vitales, para determinar si los pacientes tienen un riesgo inmediato de morir si no reciben un tratamiento agresivo. Numerosos modelos se han desarrollado en los últimos años para ayudar a predecir la mortalidad del paciente en la UCI, en función de diversos factores de salud durante su estadía. Sin embargo, estos modelos tienen inconvenientes de rendimiento. Un tipo común de modelo “global” se genera para una sola gran población de pacientes. Estos pueden funcionar bien en promedio, pero mal en algunas subdivisiones de pacientes. Por otro lado, otro tipo de modelo analiza diferentes subdivisiones como, por ejemplo, aquellas agrupadas por condiciones similares, edades de pacientes o departamentos hospitalarios, pero a menudo tienen datos limitados para capacitación y pruebas. En un artículo presentado recientemente en la conferencia Proceedings of Knowledge Discovery and Data Mining, los investigadores del MIT describen un modelo de aprendizaje automático que funciona como el mejor de ambos mundos, ya que se entrena específicamente en subdivisiones de pacientes, pero también comparte datos en todas las subdivisiones para mejorar predicciones Al hacerlo, el modelo puede predecir mejor el riesgo de mortalidad de un paciente durante sus primeros dos días en la UCI, en comparación con los modelos estrictamente globales y otros. El modelo primero recopila datos fisiológicos en registros electrónicos de salud de pacientes ingresados en la UCI, algunos de los cuales habían muerto durante su estadía. Al hacerlo, aprende altos predictores de mortalidad, como frecuencia cardíaca baja, presión arterial alta y varios resultados de pruebas de laboratorio, incluyendo altos niveles de glucosa y recuento de glóbulos blancos, entre otros, durante los primeros días y dividiendo a los pacientes en subdivisiones basadas en su estado de salud. Cuando se ingresa un nuevo paciente, el modelo puede ver los datos fisiológicos de ese paciente de las primeras 24 horas y, utilizando lo que se aprende a través del análisis de esas subdivisiones de pacientes, puede estimar mejor la probabilidad de que el nuevo paciente también muera en las siguientes 48 horas.
Además, los investigadores descubrieron que evaluar, probar y validar el modelo mediante subdivisiones específicas también destacaba las disparidades de rendimiento de los modelos globales en la predicción de la mortalidad en las subdivisiones de pacientes. Esta es una información importante para desarrollar modelos que puedan funcionar con mayor precisión con pacientes específicos. “Las UCI tienen un ancho de banda muy alto, con muchos pacientes“, dice Harini Suresh, que participó en el estudio y que es un estudiante graduado en el Laboratorio de Ciencias de la Computación e Inteligencia Artificial (CSAIL). “Es importante determinar con anticipación qué pacientes están realmente en riesgo y necesitan más atención inmediata“. Una innovación clave del trabajo es que, durante el entrenamiento, el modelo separa a los pacientes en subdivisiones distintas, que capturan aspectos del estado general de los riesgos de salud y mortalidad de un paciente. Lo hace calculando una combinación de datos fisiológicos, desglosados por hora. Los datos fisiológicos incluyen, por ejemplo, niveles de glucosa, potasio y nitrógeno, así como frecuencia cardíaca, pH sanguíneo, saturación de oxígeno y frecuencia respiratoria. Los aumentos en la presión arterial y los niveles de potasio, un signo de insuficiencia cardíaca, pueden indicar problemas de salud en otras subdivisiones. Luego, el modelo emplea un método multitarea para aprender a construir modelos predictivos. Cuando los pacientes se dividen en subdivisiones se asignan modelos sintonizados de manera diferente a cada subdivisión. Cada modelo variante puede hacer predicciones más precisas para su grupo personalizado de pacientes. Este enfoque también permite que el modelo comparta datos entre todas las subdivisiones cuando hace predicciones. Cuando se le da un nuevo paciente, hará coincidir los datos fisiológicos del paciente con todas las subdivisiones, con lo que encontrará el mejor ajuste y luego estimará mejor el riesgo de mortalidad a partir de ahí. “Estamos utilizando todos los datos del paciente y compartiendo información entre las poblaciones donde es relevante“, dice Harini Suresh. “De esta manera, podemos no sufrir problemas de escasez de datos, al tiempo que tenemos en cuenta las diferencias entre las diferentes subdivisiones de pacientes”.
“Los pacientes ingresados en la UCI a menudo difieren en por qué están allí y cómo es su estado de salud. Debido a esto, serán tratados de manera muy diferente“, agrega la estudiante graduada de CSAIL Jen Gong. Las ayudas clínicas para la toma de decisiones “deberían tener en cuenta la heterogeneidad de estas poblaciones de pacientes y asegurarse de que haya suficientes datos para predicciones precisas“. Una idea clave de este método, dice Gong, proviene del uso de un enfoque multitarea para evaluar también el rendimiento de un modelo en subdivisiones específicas. Los modelos globales a menudo se evalúan en el rendimiento general, básicamente en poblaciones completas de pacientes. Pero los experimentos de los investigadores mostraron que estos modelos realmente tienen un rendimiento inferior en las subdivisiones. El modelo global probado en el documento predijo la mortalidad en general con bastante precisión, pero disminuyó varios puntos porcentuales de precisión cuando se probó en subdivisiones individuales. “Tales disparidades de rendimiento son difíciles de medir sin evaluar por subdivisiones“, dice Gong: “Queremos evaluar qué tan bien funciona nuestro modelo, no solo en una cohorte completa de pacientes, sino también cuando lo desglosamos para cada cohorte con diferentes características médicas. Eso puede ayudar a los investigadores a mejorar la capacitación y evaluación de modelos predictivos”. Los investigadores probaron su modelo utilizando datos de la Base de datos de cuidados críticos MIMIC, que contiene decenas de datos sobre poblaciones de pacientes heterogéneas. De alrededor de 32.000 pacientes en el conjunto de datos, más de 2.200 murieron en el hospital. Utilizaron el 80 por ciento del conjunto de datos para entrenar, y el 20 por ciento para probar el modelo. Al utilizar los datos de las primeras 24 horas, el modelo agrupaba a los pacientes en subdivisiones con importantes diferencias clínicas. Dos subdivisiones, por ejemplo, contenían pacientes con presión arterial elevada durante las primeras horas, pero una disminuyó con el tiempo, mientras que la otra mantuvo la elevación durante todo el día. Esta subdivisión tuvo la tasa de mortalidad más alta. Usando esas subdivisiones, el modelo predijo la mortalidad de los pacientes durante las siguientes 48 horas con alta especificidad y sensibilidad, y varias otras métricas. El modelo multitarea superó significativamente a un modelo global en varios puntos porcentuales. A continuación, los investigadores apuntan a utilizar más datos de registros de salud electrónicos, como los tratamientos que reciben los pacientes. También esperan, en el futuro, entrenar el modelo para extraer palabras clave de notas clínicas digitalizadas y otra información. El trabajo fue apoyado por los Institutos Nacionales de Salud norteamericanos.
También la IA puede utilizarse para predecir el cáncer de mama y personalizar la atención médica. El modelo de aprendizaje profundo basado en imágenes del MIT / MGH puede predecir el cáncer de mama con hasta cinco años de anticipación. Adam Conner-Simons y Rachel Gordon, del CSAIL, nos dicen que a pesar de los grandes avances en genética e imágenes de mamografías modernas, el diagnóstico sorprende a la mayoría de las pacientes con cáncer de mama. Para algunos, llega demasiado tarde. El diagnóstico posterior significa tratamientos agresivos , resultados inciertos y más gastos médicos. Como resultado, la identificación de pacientes ha sido un pilar central de la investigación del cáncer de mama y la detección temprana efectiva. Con eso en mente, un equipo del Laboratorio de Ciencias de la Computación e Inteligencia Artificial (CSAIL) del MIT y el Hospital General de Massachusetts (MGH) ha creado un nuevo modelo de aprendizaje profundo que puede predecir, a partir de una mamografía, si es probable que una paciente desarrolle tanto cáncer de mama en cinco años en el futuro. Entrenado en mamografías y resultados conocidos de más de 60.000 pacientes del Hospital General de Massachusetts, el modelo aprendió los patrones sutiles en el tejido mamario que son precursores de tumores malignos. La profesora del MIT Regina Barzilay, una sobreviviente de cáncer de mama, dice que la esperanza es que sistemas como estos permitan a los médicos personalizar los programas de detección y prevención a nivel individual, haciendo del diagnóstico tardío una reliquia del pasado. Aunque se ha demostrado que la mamografía reduce la mortalidad por cáncer de mama, existe un debate continuo sobre la frecuencia con la que se realiza el examen y cuándo comenzar. Mientras que la Sociedad Estadounidense del Cáncer recomienda un examen anual a partir de los 45 años, el Grupo de trabajo preventivo de Estados Unidos recomienda un examen cada dos años a partir de los 50 años. “En lugar de adoptar un enfoque único para todos, podemos personalizar la detección según el riesgo de una mujer de desarrollar cáncer“, dice Barzilay , autora principal de un nuevo artículo sobre el proyecto que se presentó en Radiología. “Por ejemplo, un médico podría recomendar que un grupo de mujeres se haga una mamografía cada dos años, mientras que otro grupo de mayor riesgo podría hacerse un examen de resonancia magnética complementario“. Barzilay es profesor de Delta Electronics en el CSAIL y del Departamento de Ingeniería Eléctrica e Informática del MIT, así como miembro del Instituto Koch para la Investigación Integral del Cáncer en el MIT.
El modelo del equipo fue significativamente mejor para predecir el riesgo que los enfoques existentes. Colocó con precisión al 31% de todos los pacientes con cáncer en su categoría de mayor riesgo, en comparación con solo el 18% de los modelos tradicionales. La profesora de Harvard, Constance Lehman, profesora de radiología en la Escuela de Medicina de Harvard y jefe de división de imágenes mamarias en el Hospital General de Massachusetts, dice que anteriormente la comunidad médica había brindado un apoyo mínimo para las estrategias de detección basadas en el riesgo y no en la edad. “Esto se debe a que antes no teníamos herramientas precisas de evaluación de riesgos que funcionaran para mujeres individuales. Nuestro trabajo es el primero en demostrar que es posible“. Barzilay y Lehman escribieron conjuntamente el documento junto al autor principal, Adam Yala , un estudiante de doctorado del CSAIL. Otros coautores del MIT incluyen la estudiante de doctorado Tal Schuster y la ex estudiante de maestría Tally Portnoi. Desde el primer modelo de riesgo de cáncer de mama de 1989, ha sido impulsado el desarrollo en gran medida por el conocimiento humano y la intuición de cuáles son los principales factores de riesgo, como la edad, los antecedentes familiares de cáncer de mama y de ovario, los factores hormonales y reproductivos y la densidad mamaria. Sin embargo, la mayoría de estos marcadores solo se correlacionan débilmente con el cáncer de mama. Como resultado, estos modelos aún no son muy precisos a nivel individual, y muchas organizaciones continúan pensando que los programas de detección basados en el riesgo no son posibles, dadas esas limitaciones. En lugar de identificar manualmente los patrones en una mamografía que pueden conducir a un cáncer futuro, el equipo de MIT / MGH entrenó un modelo de aprendizaje profundo para deducir los patrones directamente de los datos. Usando información de más de 90.000 mamografías, el modelo detectó patrones demasiado sutiles para que el ojo humano los detectase. Según Lehman: “Desde la década de 1960, los radiólogos han notado que las mujeres tienen patrones únicos y ampliamente variables de tejido mamario visibles en la mamografía. Estos patrones pueden representar la influencia de la genética, las hormonas, el embarazo, la lactancia, la dieta, la pérdida de peso y el aumento de peso. Ahora podemos aprovechar esta información detallada para ser más precisos en nuestra evaluación de riesgos a nivel individual“.
El proyecto también tiene como objetivo hacer que la evaluación de riesgos sea más precisa para las minorías raciales, en particular. Muchos de los primeros modelos se desarrollaron en poblaciones blancas y fueron mucho menos precisos para otras razas. El modelo MIT / MGH, por su parte, es igualmente preciso para las mujeres blancas y negras. Esto es especialmente importante dado que se ha demostrado que las mujeres negras tienen un 42% más de probabilidades de morir de cáncer de mama debido a una amplia gama de factores que pueden incluir diferencias en la detección y el acceso a la atención médica. Según Allison Kurian, profesora asociada de medicina e investigación y política de salud en la Facultad de Medicina de la Universidad de Stanford: “Es particularmente sorprendente que el modelo funcione igual de bien para las personas blancas y negras, lo que no ha sido el caso con las herramientas anteriores. Si se valida y se pone a disposición para un uso generalizado, esto realmente podría mejorar nuestras estrategias actuales para estimar el riesgo“. Barzilay dice que su sistema también podría algún día permitir que los médicos usen mamografías para ver si las pacientes tienen un mayor riesgo con otros problemas de salud, como enfermedades cardiovasculares u otros tipos de cáncer. Los investigadores están ansiosos por aplicar los modelos a otras enfermedades y dolencias, y especialmente a aquellos con modelos de riesgo menos efectivos, como el cáncer de páncreas. Adam Yala nos dice que: “Nuestro objetivo es hacer que estos avances sean parte del estándar de atención. Al predecir quién desarrollará cáncer en el futuro, esperamos poder salvar vidas y contraer cáncer antes de que surjan los síntomas“.
Según Adam Conner-Simons, del CSAIL, un modelo desarrollado en el Laboratorio de Ciencias de la Computación e Inteligencia Artificial del MIT podría reducir los falsos positivos y las cirugías innecesarias relacionadas con el cáncer de mama. Cada año, 40.000 mujeres mueren de cáncer de mama solo en los Estados Unidos. Cuando los cánceres se detectan temprano, a menudo se pueden curar. Las mamografías son la mejor prueba disponible, pero siguen siendo imperfectas y a menudo dan como resultados falsos positivos que pueden conducir a biopsias y cirugías innecesarias. Una causa común de falsos positivos son las llamadas lesiones de “alto riesgo” que parecen sospechosas en las mamografías y tienen células anormales cuando se analizan mediante biopsia con aguja. En este caso, el paciente generalmente se somete a una cirugía para extirpar la lesión. Sin embargo, las lesiones resultan benignas en la cirugía el 90% de las veces. Esto significa que cada año miles de mujeres pasan por cirugías dolorosas, caras e inductoras de cicatrices que ni siquiera eran necesarias. La pregunta es, ¿cómo se pueden eliminar las cirugías innecesarias mientras se mantiene el importante papel de la mamografía en la detección del cáncer? Investigadores del Laboratorio de Ciencias de la Computación e Inteligencia Artificial (CSAIL) del MIT, el Hospital General de Massachusetts y la Escuela de Medicina de Harvard creen que la respuesta consiste en recurrir a la inteligencia artificial (IA). Como primer proyecto para aplicar IA para mejorar la detección y el diagnóstico, los equipos colaboraron para desarrollar un sistema de IA que utiliza el aprendizaje automático para predecir si una lesión de alto riesgo, identificada en la biopsia con aguja después de una mamografía, se convertirá en cáncer en la cirugía. Cuando se probó en 335 lesiones de alto riesgo, el modelo diagnosticó correctamente el 97% de los cánceres de mama como malignos y redujo el número de cirugías benignas en más del 30% en comparación con los enfoques actualmente existentes. Según Regina Barzilay , profesora de ingeniería eléctrica y ciencias de la computación Delta Electronics del MIT, y una sobreviviente de cáncer de mama: “Debido a que las herramientas de diagnóstico son tan inexactas, existe una tendencia comprensible para que los médicos hagan un examen de detección de cáncer de mama. Cuando existe tanta incertidumbre en los datos, el aprendizaje automático es exactamente la herramienta que necesitamos para mejorar la detección y evitar un exceso en el tratamiento“.
Entrenado en la información sobre más de 600 lesiones existentes de alto riesgo, el modelo de IA busca patrones entre muchos elementos de datos diferentes que incluyen datos demográficos, antecedentes familiares, biopsias pasadas e informes de patología. Constance Lehman, profesora de la Facultad de Medicina de Harvard y responsable de Breast Imaging en el Departamento de Radiología del Hospital General de Massachusetts, nos dice: “Hasta donde sabemos, este es el primer estudio que aplica el aprendizaje automático a la tarea de distinguir las lesiones de alto riesgo que necesitan cirugía de las que no lo necesitan. Creemos que esto podría ayudar a las mujeres a tomar decisiones más informadas sobre su tratamiento, y que podríamos proporcionar enfoques más específicos para la atención médica en general“. Cuando una mamografía detecta una lesión sospechosa, se realiza una biopsia con aguja para determinar si es cáncer. Aproximadamente el 70% de las lesiones son benignas, el 20% son malignas y el 10% son lesiones de alto riesgo. Los médicos manejan las lesiones de alto riesgo de diferentes maneras. Algunos realizan cirugía en todos los casos, mientras que otros realizan cirugía solo para lesiones que tienen tasas de cáncer más altas, como “hiperplasia ductal atípica” (ADH) o un “carcinoma lobular in situ” (LCIS). El primer enfoque requiere que el paciente se someta a una cirugía dolorosa, lenta y costosa que generalmente es innecesaria; El segundo enfoque es impreciso y podría dar como resultado la ausencia de cánceres en lesiones de alto riesgo que no sean ADH y LCIS. Según Manisha Bahl, del Departamento de Radiología del Hospital General de Massachusetts: “La gran mayoría de los pacientes con lesiones de alto riesgo no tienen cáncer, y estamos tratando de encontrar los pocos que sí lo tienen. En un escenario como este, siempre existe el riesgo de que cuando se intente aumentar la cantidad de cánceres que pueda identificar, también aumente la cantidad de falsos positivos que encuentre“. Usando un método conocido como un “clasificador de bosque aleatorio“, el modelo del equipo dio como resultado menos cirugías innecesarias en comparación con la estrategia de realizar siempre una cirugía, al tiempo que también fue capaz de diagnosticar más lesiones cancerosas que la estrategia de realizar solo una cirugía tradicional. Específicamente, el nuevo modelo diagnostica el 97% de los cánceres en comparación con el 79%.
Marc Kohli, director de informática clínica en el Departamento de Radiología e Imagen Biomédica de la Universidad de California en San Francisco, nos dice: “Este trabajo destaca un ejemplo en el uso de tecnología de vanguardia de aprendizaje automático para evitar cirugías innecesarias. Este es el primer paso hacia la comunidad médica que adopta el aprendizaje automático como una forma de identificar patrones y tendencias que de otro modo serían invisibles para los humanos“. Constance Lehman dice que los radiólogos del Hospital General de Massachusetts comenzarán a incorporar pronto el modelo en su práctica clínica: “En el pasado, podríamos haber recomendado que todas las lesiones de alto riesgo se extirparan quirúrgicamente. Pero ahora, si el modelo determina que la lesión tiene una probabilidad muy baja de ser cancerosa en un paciente específico, podemos tener una discusión más informada con nuestro paciente sobre sus opciones. Puede ser razonable que a algunos pacientes les sigan las lesiones con imágenes en lugar de extirparlas quirúrgicamente“. El equipo dice que todavía están trabajando para perfeccionar aún más el modelo. “En el trabajo futuro esperamos incorporar las imágenes reales de las mamografías y las imágenes de las diapositivas de la patología, así como información más extensa de los pacientes de los registros médicos“, dice Bahl. En el futuro, el modelo también podría modificarse fácilmente para aplicarse a otros tipos de cáncer e incluso a otras enfermedades por completo. Según Barzilay: “Un modelo como este funcionará cada vez que tenga muchos factores diferentes que se correlacionan con un resultado específico. Esperamos que nos permita comenzar a ir más allá de un enfoque único para el diagnóstico médico“. En fin, como he intentado mostrar en este artículo, la irrupción de la Inteligencia Artificial en el campo médico parece realmente imparable. Esperemos que su utilización sea siempre para favorecer a los seres humanos.
Fuentes:
- Stuart Russell y Peter Norvig – Inteligencia artificial: Un enfoque moderno
- Alejandro Madruga – Inteligencia artificial, el futuro del hombre
- Alberto García Serrano – Inteligencia Artificial. Fundamentos, práctica y aplicaciones
- Margaret Boden y Inmaculada Pérez Parra – Inteligencia artificial
- Ray Kurzweil – La Singularidad está cerca: Cuando los humanos transcendamos la biología
- Steve Hanley (http://news.mit.edu/) – Artificial Intelligence Algorithm Creates Antibiotic Effective Against Drug Resistant Bacteria
- Stefanie Koperniak (http://news.mit.edu/) – Applying machine learning to challenges in the pharmaceutical industry
- Will Douglas Heaven (http://news.mit.edu/) – A neural network can help spot Covid-19 in chest X-rays
- Will Douglas Heaven (http://news.mit.edu/) – AI could help with the next pandemic—but not with this one
- Anne Trafton (http://news.mit.edu/) – Artificial intelligence yields new antibiotic
- Rob Matheson (http://news.mit.edu/) – Cryptographic protocol enables greater collaboration in drug discovery
- Kim Martineau (http://news.mit.edu/) – Democratizing artificial intelligence in health care
- Maria Iacobo (http://news.mit.edu/) – Gift will allow MIT researchers to use artificial intelligence in a biomedical device
- Becky Ham (http://news.mit.edu/) – Guided by AI, robotic platform automates molecule manufacture
- Kim Martineau (http://news.mit.edu/) – Q&A: Markus Buehler on setting coronavirus and AI-inspired proteins to music
- Rob Matheson (http://news.mit.edu/) – Model improves prediction of mortality risk in ICU patients
- Rob Matheson (http://news.mit.edu/) – Model learns how individual amino acids determine protein function
- Karen Hao (http://news.mit.edu/) – Over 24,000 coronavirus research papers are now available in one place
- Jo Marchant (http://news.mit.edu/) – Powerful antibiotics discovered using AI
- Karen Hao (http://news.mit.edu/) – This is how the CDC is trying to forecast coronavirus’s spread
- David L. Chandler (http://news.mit.edu/) – Translating proteins into music, and back
- Adam Conner-Simons and Rachel Gordon (http://news.mit.edu/) – Using AI to predict breast cancer and personalize care
- Adam Conner-Simons (http://news.mit.edu/) – Using artificial intelligence to improve early breast cancer detection
- Gina Vitale (http://news.mit.edu/) – Using machine learning for medical solutions
¿Cómo puede la Inteligencia artificial ayudar a combatir pandemias como el Covid-19?